|
Pianeta
Chimica.it
Responsabile:
Prof. Mauro Tonellato
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|

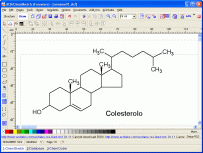
Chemsketch
Disegna molecole in 2D e le trasforma in 3D
Assegna il nome IUPAC
Chemsketch (distribuito gratuitamente dalla ACD Labs) è
il programma più semplice ed immediato per disegnare molecole
e generare il loro nome IUPAC (se sono composte da meno di
50 atomi.
Fornisce anche alcuni dati sulla molecola disegnata come peso molecolare,
composizione percentuale, formula bruta, indice di rifrazione, densità,
tensione superficiale, ecc.
Ma questo è solo il
primo passo...
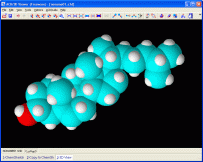
Permette di generare il modello tridimensionale ottimizzato
della molecola (vedi foto qui a lato) e di modificare lunghezze
e angoli di legame, come pure angoli di torsione e
configurazioni R/S!
https://www.acdlabs.com/resources/free-chemistry-software-apps/chemsketch-freeware/
Provate ad usare Chemsketch in classe!
(Lezione di Chimica al Computer con Chemsketch)
La dispensa Nomenclatura IUPAC oltre
a spiegare nei dettagli la nomenclatura, è una guida per
imparare ad usare Chemsketch per imparare meglio la nomenclatura.
 Nomenclatura
IUPAC
Nomenclatura
IUPAC
|
|
|
|
|
|
|
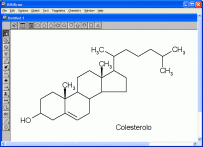
ISIS Draw 2.5
ServicePack4 (compatibile con Win7)
Disegna molecole in 2D
Tra i molti programmi per disegnare molecole
in 2D, forse il migliore è ISIS Draw 2.5 (gratuito!)
distribuito da MDL. Si tratta di un programma completo ed efficace
che permette in pochi secondi di disegnare molecole anche complesse
dall'aspetto professionale per inserirle in dispense o appunti
personali. (vedi foto qui a lato). Tutti le molecole nelle dispense
di pianetachimica sono state disegnate con ISIS Draw
Dato che non è più distribuito da MD, lo potete
scaricare qui
ISIS Draw 2.5
N.B. Può essere usato anche con Windows 7
a patto di impostare la compatibilità con WinXP (pulsante
destro - proprietà - compatibilità - Win XP)
Non è compatibile con Widows 10. Con questo sistema
operativo dovete usare la nuova versione Biovia Draw 2018 descritto
qui sotto.
BioviaDraw
Disegna molecole
in 2D
Assegna il nome IUPAC
https://www.3dsbiovia.com/products/collaborative-science/biovia-draw/
E' la nuova versione di ISISDraw compatibile con windows 10. E'
un buon prodotto efficiente e affidabile compatibile con i vecchi
file di ISISDraw. E' distribuito gratuitamente per insegnanti
e studenti.
|
|
|
|
|
|
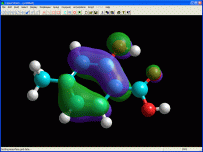
ArgusLab 4.0
Disegna e ottimizza molecole
in 3D,
mostra gli orbitali molecolari, calcola le energie di formazione
Con questo ottimo programma distribuito (gratuitamente)
da Mark Thompson della Planaria Software, è possibile sia
creare sia importare molecole per esaminarle in 3 dimensioni con
un'ottima qualità grafica.
http://www.arguslab.com
Inoltre sono disponibili delle funzioni avanzate di calcolo per
l'ottimizzazione tridimensionale delle molecole, per il calcolo
della superficie degli orbitali HOMO e LUMO che permettono
di indagare la reattività della molecola (vedi foto
qui a lato).
|
|
|
|
|
|
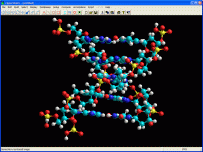
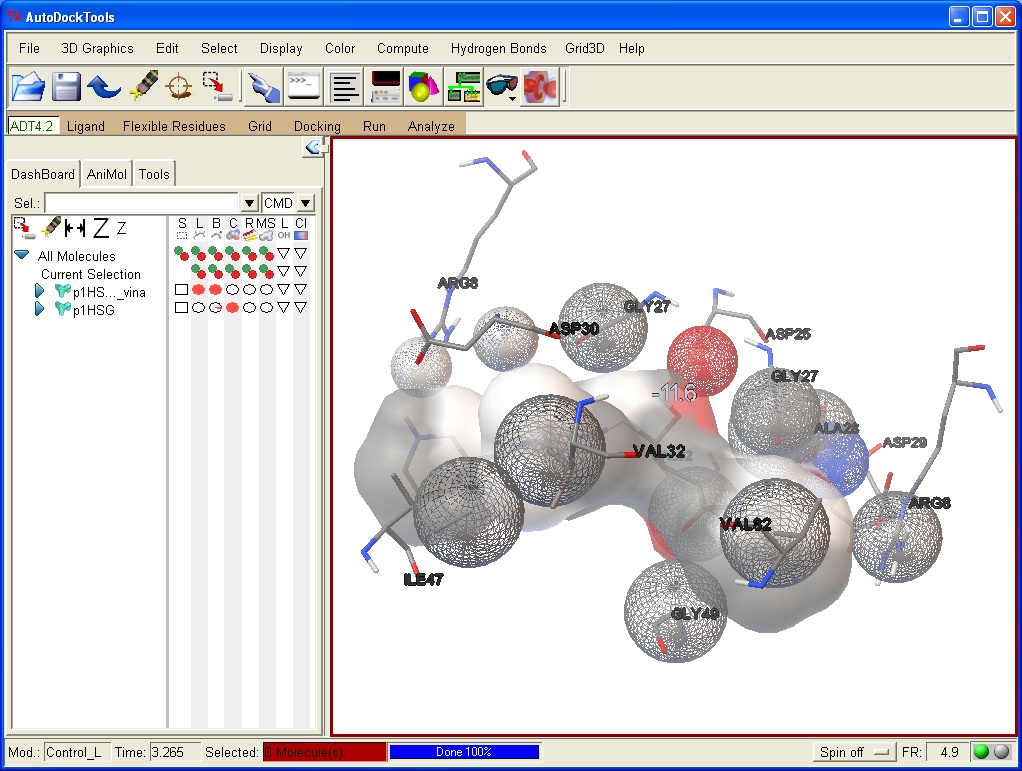
MGL Tools e AutoDock Vina
MGL Tools http://mgltools.scripps.edu/downloads
AutoDock Vina http://vina.scripps.edu/download.html
MGL Tools è
un programma di modellistica molecolare ideale per le proteine.
E' anche ideale per elaborare le proteine e i legandi prima di eseguire
il docking molecolare con AutoDock Vina. Permette inoltre di valutare
i risultati del docking. E' stato sviluppato al MGL Molecular Graphic
Laboratory dello Scripps Research Institute
AutoDock Vina è il
programma di docking molecolare più usato al mondo. E' stato
sviluppato dal
Dr. Oleg Trott del The Scripps Research Institute. E' un programma
molto avanzato che sfrutta al massimo la potenza di calcolo delle
macchine multiprocessore e consente non solo di esplorare le varie
configurazioni del legando, ma può anche trattare come flessibili
i residui degli amminoacidi del sito attivo.
|
|
|
|
|
|
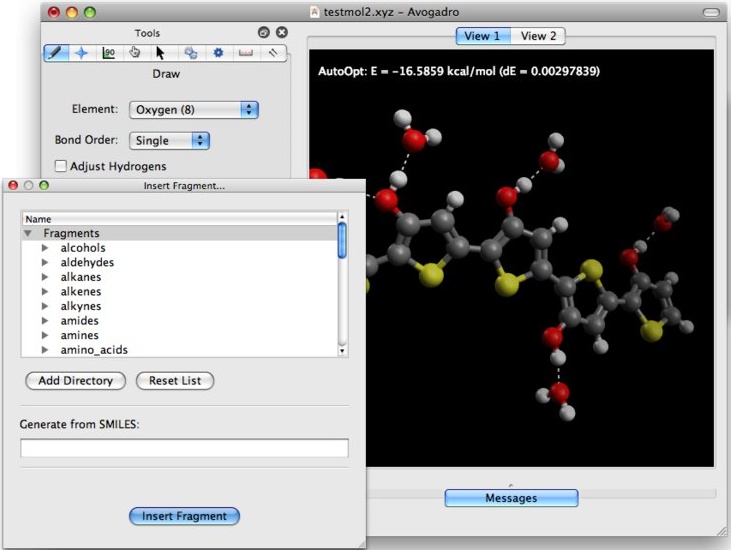
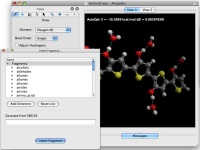
Avogadro 1.2
https://avogadro.cc/
Avogadro è un programma di modellistica molecolare che permette
di costruire molecole organiche anche complesse, di calcolarne la
struttura più stabile con tecniche di meccanica molecolare
come UFF e MMFF94 e fornisce anche immagini di ottima qualità
grafica. Permette inoltre di aggiungere idrogeni alle molecole ottenendo
diversi gradi di protonazione in funzione del pH scelto.
Avogadro può essere usato insieme con programmi di chimica
computazionale come ORCA
(o Gaussian, a pagamento) e così permette di ottimizzare
le molecole anche con metodi quantomeccanici avanzati, calcolare
energia e forma degli orbitali molecolari, calcolare le frequenze
di assorbimento IR, valutare gli stati di transizione delle reazioni
ecc. Davvero notevole!
ORCA
(https://orcaforum.kofo.mpg.de/app.php/portal)
Pacchetto software open-source per eseguire calcoli avanzati di
chimica quantistica sulle molecole.
La sua interfaccia grafica naturale è Avogadro.
Distribuito gratuitamente per scopi didattici da un gruppo del Max
Plank Institute.
Nel Forum di ORCA è possibile scaricare una versione aggiornata
di Avogadro dedicato ad ORCA.
|
|
|
|
|
|
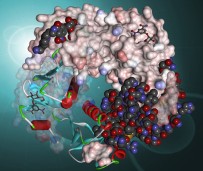
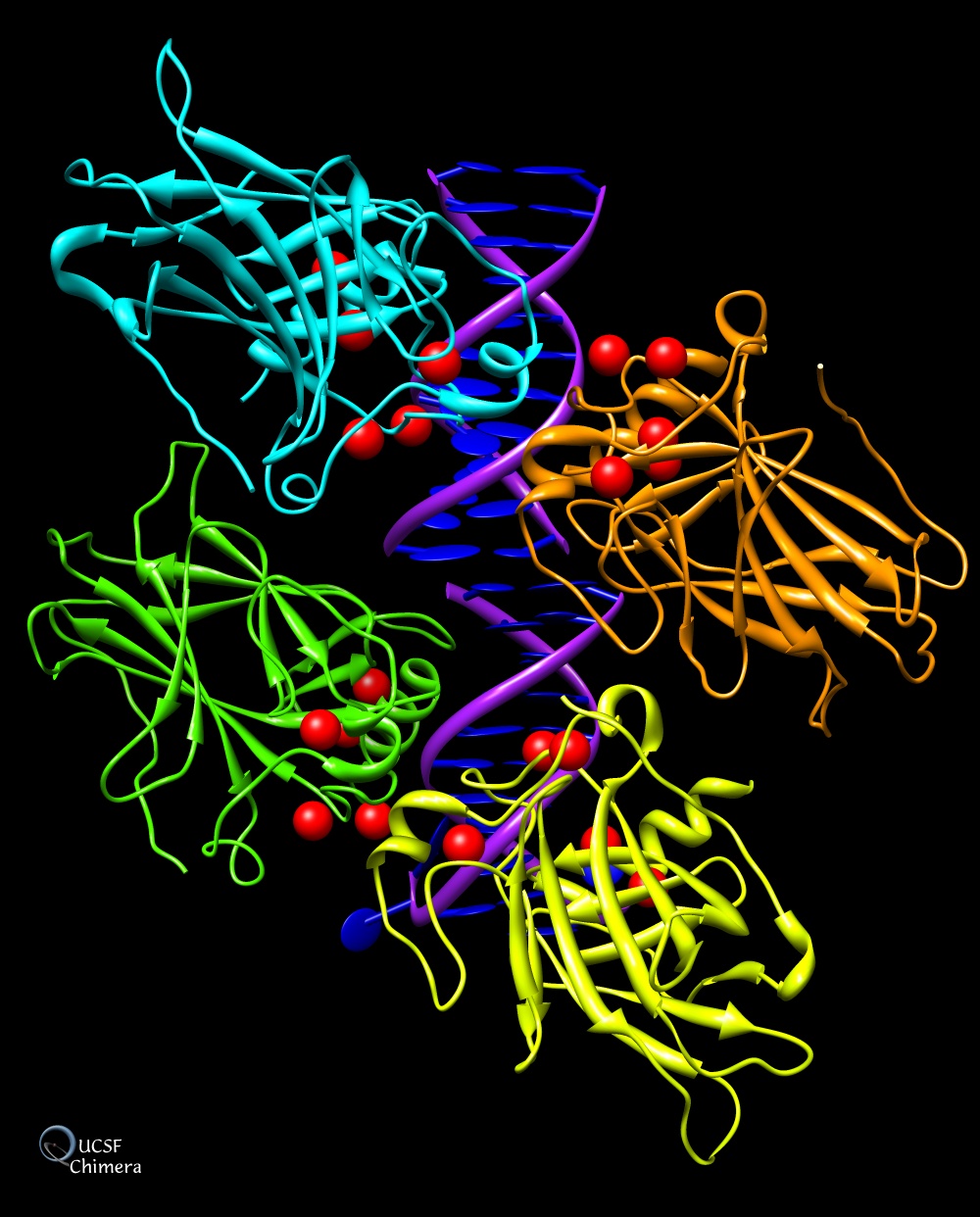
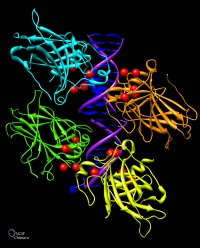
Chimera
https://www.cgl.ucsf.edu/chimera/
UCSF Chimera è un ottimo programma per visualizzare e analizzare
la struttura di molecole e macromolecole. E' adatto anche a visualizzare
il risultati del docking molecolare. Permette di generare immagini
di grande qualità grafica con una buona resa di luci, ombre,
riflessi e colori. Nel sito ufficiale si trova una galleria di immagini
e una documentazione completa, nonchè alcuni tutorial. Può
essere usato gratuitamente per uso personale o didattico.
Molte delle immagini delle molecole del mese sono state realizzate
con Chimera
|
|
|
|
|
|
|
gNMR 4.1
Realizza spettri NMR
simulati
Questa è la versione più
vecchia del programma gNMR, ma è più semplice ed
immediato da usare rispetto alla nuova versione 5.06 (vedi qui
sotto).
L'autore del programma, il prof. Budzelaa, mi ha autorizzato a
distribuirlo direttamente precisando che lui NON intende più
sostenerlo o fornire assistenza per questa vecchia versione.
Il programma è scaricabile direttamente dal link qui
sotto
Per generare lo spettro, è necessario disegnare prima la
molecola con Isis Draw e poi importare il file generato (.skc)
in gNMR. (File / Import / MDL-SKC). L'unica funzione che non è
inclusa nell versione demo è il copia e incolla degli spettri
che così non si possono esportare direttamente in altri
programmi (per esempio Word).
Questo problema si supera facilmente premendo il pulsante "stampa
schermo" sulla tastiera del computer. L'immagine dello schermo
si può incollare in programmi di grafica come l'ottimo
Irfanview (free) che consente di ritagliare lo spettro con la
funzione Edit \ Crop selection.
gNMR 4.1 si può scaricare QUI premendo
il pulsante destro del mouse sul seguente link:
gNMR 4.1 download
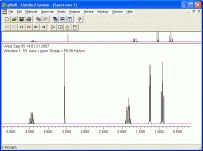
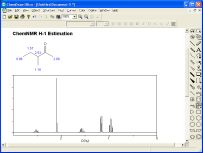
gNMR 5.06
Realizza spettri NMR simulati
La spettroscopia NMR è una tecnica molto potente per l'indagine
strutturale di molecole organiche. Purtroppo però uno strumento
NMR ha un costo così elevato che è fuori dalla portata
di una scuola superiore.
Gli studenti possono esercitarsi con la spettroscopia NMR usando
gNMR 5.06 (gratuito!), di P.H.M. Budzelaar e distribuito
dalla Ivorysoft, un programma che calcola lo spettro NMR di una
molecola assegnata. Nella foto qui a lato si può vedere
lo spettro del 2-butanolo. Rispetto alla precedente versione 4.1
il programma è ora utilizzabile gratuitamente per la
didattica nella sua versione completa. E' stata migliorata
l'accuratezza di previsione dei chemical shift.
http://home.cc.umanitoba.ca/~budzelaa/gNMR/gNMR.html
http://home.cc.umanitoba.ca/~budzelaa/gNMR/gNMR-license.html
Durante l'installazione del programma viene
chiesto il numero di serie, utilizzare N500-011292-715.
Per generare lo spettro, è necessario disegnare prima la
molecola con Isis Draw e poi importare il file generato (.skc)
in gNMR. (File / Import / MDL-SKC).
N.B. : i gruppi ossidrili non vanno disegnati come OH ma come
O-H con il legame esplicitato. Così pure per i gruppi amminici,
ecc.
Per vedere una raccolta di spettri NMR generati con gNMR vai a
Spettroscopia NMR.
|
|
|
|
|
|
|
Orbital Viewer
Mostra gli orbitali di atomi e molecole
Con questo programma si possono
rappresentare tutti gli orbitali atomici per meglio comprenderne
la forma e anche la struttura interna compresi i nodi. Si possono
inoltre avvicinare due o più atomi fino ad ottenere molecole.
Il programma è disponibile gratuitamente al sito:
http://www.orbitals.com/orb/ov.htm
|
|
|
|
|
|
|
Biovia Discovery Studio Visualizer
Visualizza le molecole in
3D
Tra i programmi che permettono di vedere e manipolare le molecole
in tre dimensioni, va segnalato anche Biovia DS Visualizer
(gratuito!!) che oltre alle molecole normali è particolarmente
adatto a manipolare proteine, DNA e RNA.
DS Visualizer si rivela potente e flessibile nello scegliere ed
evidenziare ogni parte della molecola, offre molte opzioni di
visualizzazione e produce immagini di buona qualità grafica.
https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/visualization.html
|
|
|
|
|
|
ChemOffice Professional
Disegna molecole in 2D, assegna il nome IUPAC,
calcola costanti chimico-fisiche, genera spettri NMR
Questo è un programma professionale che
può essere provato gratuitamente per 15 giorni scaricandolo
al sito:
http://www.cambridgesoft.com/Ensemble_for_Chemistry/ChemOffice/
Svolge le funzioni di più programmi gratuiti insieme: disegna
come ISIS Draw, calcola le costanti chimico fisiche come
Chemsketch (è più completo), assegna il nome
IUPAC come Chemsketch, traccia lo spettro NMR come
gNMR (è più preciso nella previsione dei corretti
chemical shift).
Inoltre calcola la struttura 3D delle molecole, mostra
gli orbitali molecolari, esegue calcoli quantomeccanici
come ArgusLab.
|
|
|
|
|
|
|
HyperChem 8.0.10
Programma professionale di modellistica
molecolare.
Ottimo programma della Hypercube, disegna le molecole in 3D, ottimizza
la loro struttura tridimensionale calcolandone l'energia minima
utilizzando metodi di meccanica quantistica, ricava lo spettro
IR delle molecole e per ogni frequenza assorbita mostra un'animazione
delle vibrazioni molecolari. Mostra tutti gli orbitali di una
molecola, sia quelli di legame che di antilegame e in particolare
consente di valutare la struttura degli orbitali HOMO e LUMO per
meglio comprendere la reattività delle molecole. Fa anche
calcoli di termodinamica e di cinetica delle reazioni. Disegna
in pochi istanti sequenze di amminoacidi, di nucleotidi o di zuccheri.
Ha ottime capacità di rendering e offre immagini di qualità
fotografica.
Si può ottenere una copia gratuita del programma valida
per 10 giorni collegandosi al sito:
http://www.hyper.com
|
|
|
|
|
|
Altri programmi interessanti
Spartan Wavefunction ottimo programma professionale di modellistica
molecolare simile a Hyperchem. Usa metodi quantomeccanici avanzati
e calcola spettri IR, spetri NMR, stati di transizione di reazioni
e molto altro. Dispone di un database molto vasto con cui confrontare
gli spettri calcolati.
MestreNova Permette di calcolare uno spettro NMR simulato di
una molecola a piacere
Permette di trasformare un segnale FID prodotto da uno strumento
di acquisizione di spettri NMR in uno spettro NMR leggibile applicando
la Trasformata di Fourier.
Crocodile Chemistry simula un intero laboratorio chimico e permette
di realizzare centinaia di reazioni ed esperienze di laboratorio
e di seguirle osservando le variazioni di pH, temperatura, composizione,
ecc.
Swiss PDB Viewer un altro ottimo visualizzatore di macromolecole
(proteine, DNA) in formato Pdb. E' del tutto gratuito.
ConceptDraw Office v8 Disegna impianti chimici industriali.
Gaussian Programma di chimica computazionale professionale.
Ottimizza le molecole con tutte le tecniche disponibili, calcola
spettri IR, Calcola forma ed energia degli orbitali MO, Calcola
lo stato di transizione e l'energia durante la reazione, esegue
simulazioni di dinamica molecolare
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|