|
Chimica al computer con
MGL Tools / AutoDock
 Struttura delle Proteine
Struttura delle Proteine
Proteine e Legandi
Docking su COX2
Progettare
nuovi farmaci
Chimica al computer con
ArgusLab
1-butene
e 2-butene
Carbocatione
1° e 2°
Acetone
e tautomeria
Dieni
coniugati (pdf)
Benzene
e aromaticità (pdf)
Legame covalente
Conformazione alcani
Ponte Cloronio
Diels-Alder
Enolo
Mono e Disostituito (pdf)
 Chimica al Computer
Chimica al Computer
PianetaChimica home
|
|
Le proteine sono macromolecole molto complesse, la loro struttura può
essere capita più facilmente con un programma di modellistica
molecolare come MGL Tools. Questo programma mostra il modello tridimensionale
della proteina nello schermo del computer e consente di esaminarlo e
manipolarlo come se fosse un oggetto nelle nostre mani. Questo non solo
ci aiuta a capire meglio il significato di struttura primaria, secondaria,
terziaria e quaternaria delle proteine, ma ci permette di comprendere
meglio anche altre proprietà delle proteine, come l'interazione
legando - sito attivo negli enzimi o l'azione degli inibitori.
Questa lezione può essere affrontata in due modi.
1) Online. Potete leggere le istruzioni ed eseguirle passo passo
al computer con MGL Tools, aiutandovi con le illustrazioni che chiariscono
ogni passaggio.
2) In aula informatica con la classe. Se siete insegnanti di
chimica, potete adattare la lezione alle esigenze della vostra classe
e proporla in aula informatica ai vostri allievi.
La durata di questa lezione è di circa un'ora.
N.B. Cliccando sulle figure potrete vedere le immagini a pieno
schermo.

Gli argomenti di questa lezione sono:
-- Legando e sito di legame in una proteina
-- Analisi delle interazioni tra legando e sito di legame
|
|
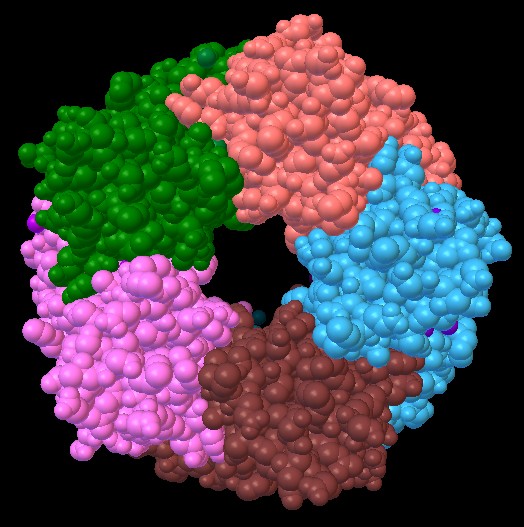
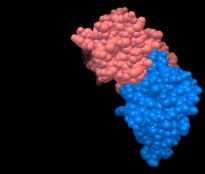
1uv6 - fig. 1
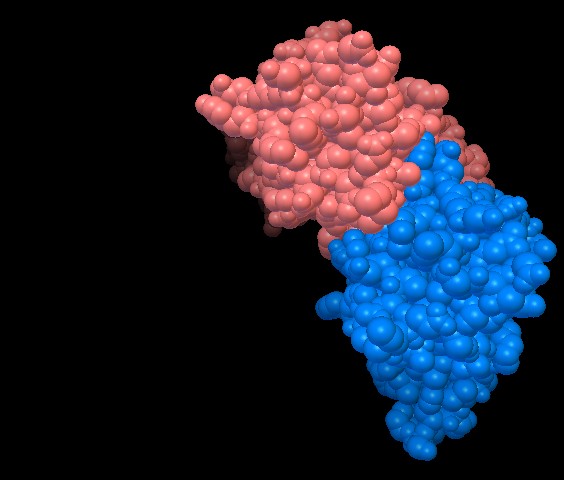
1uv6_tut - fig. 2
|
|
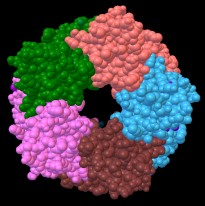
Recettore dell'acetilcolina
Anche in questa seconda lezione sulle proteine, studieremo il recettore
dell'acetilcolina, ma, invece della proteina completa (mostrata in
figura 1) formata da 5 catene, useremo 1uv6_tut (figura 2) che contiene
solo le catene C e D, e che abbiamo già usato nella lezione precedente.
In questa struttura, al posto dell'acetilcolina, è presente un
suo analogo, carbammilcolina, che si lega in modo più stabile
nella proteina e così proteina e legando possono essere cristallizzati
insieme.
Il recettore dell'acetilcolina si trova sulla superficie delle cellule
muscolari nelle sinapsi nervo-muscolo. Quando l'impulso nervoso arriva
alla fine del neurone, questo rilascia un getto di neurotrasmettitori
come l'acetilcolina, che giunge alla cellula muscolare e si lega a specifici
recettori il cui poro centrale si apre e lascia entrare un flusso di ioni
sodio che innesca la contrazione muscolare. Questi recettori svolgono
un ruolo molto delicato perchè, se vengono paralizzati nella forma
aperta, costringono i muscoli a rimanere contratti per un tempo indefinito
provocando un irrigidimento incontrollato che può portare alla
morte. Alcuni veleni tra i più pericolosi, come il veleno del
cobra e il veleno della conchiglia a cono, attaccano proprio
il recettore dell'acetilcolina.
Aprire
il file PDB
Scaricate il file 1uv6_tut.pdb
e collocatelo in una cartella che chiamerete Acetilcolina nella
root directory c:\.
Lanciate MGL Tools e cliccate File / Preferences / Set per aprire
una finestra di dialogo, quindi scrivete in Startup Directory la
posizione della cartella Acetilcolina, c:\Acetilcolina.
In MGL Tools cliccate File / Read Molecule e aprite il file 1uv6_tut.pdb.
|
|
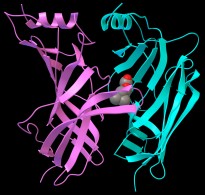
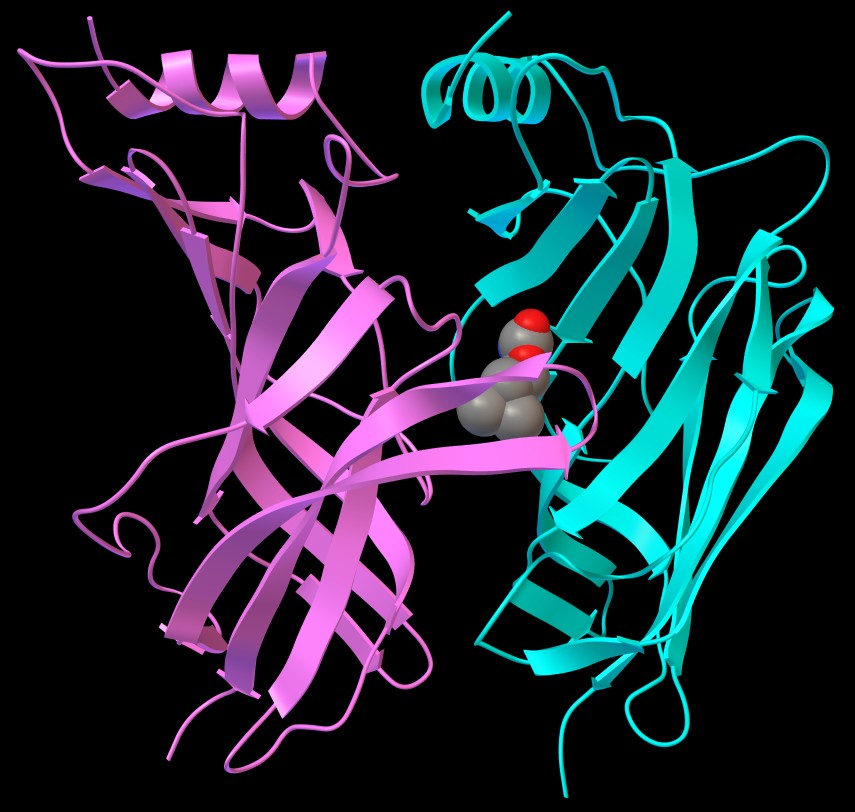
fig. 3
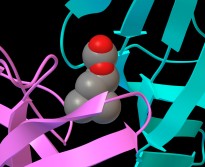
fig. 4
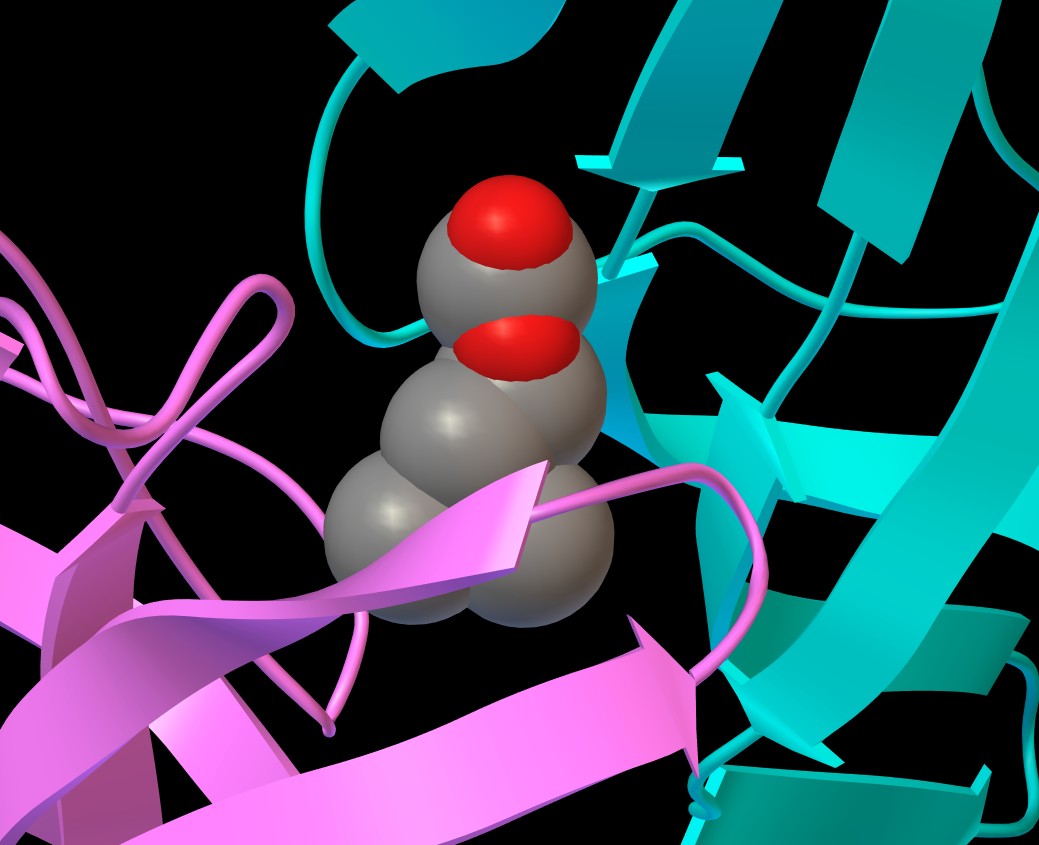
figura 5
figura 5b
|
|
Individuare il legando
Deselezionate la rappresentazione linee (colonna L) per 1uv6_tut
e scegliete Ribbon (colonna R).
Colorate la struttura secondaria di 1uv6_tut (colonna Cl) con
l'opzione By Chain.
Cliccate sul triangolino azzurro accanto a 1uv6_tut per vedere
le subunità di cui è composto (C e D)
Cliccate sul triangolino azzurro accanto alla catena C e scorrete
i residui verso il basso fino ad incontrare il legando CCE-1206
(carbammilcolina).
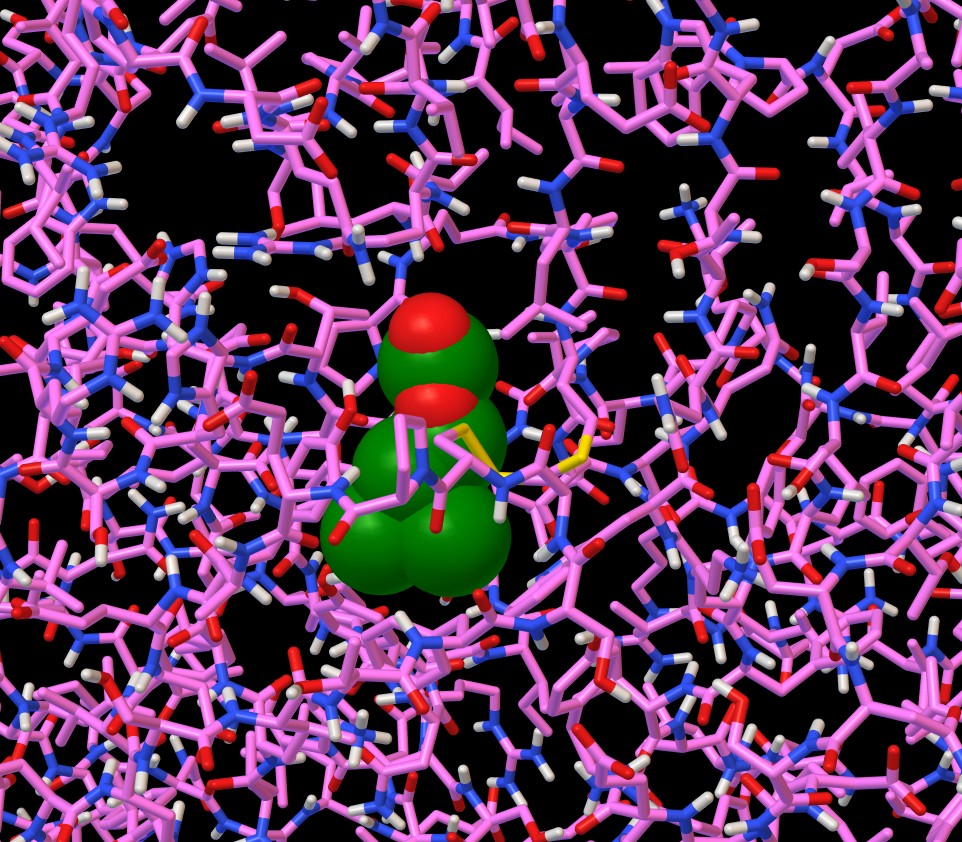
Cliccate su questa e scegliete la rappresentazione con sfere
(colonna C) e poi coloratela (colonna Cl) scegliendo cpk
e poi By atom type. Otterrete le immagini di figura 3 e figura
4 dove si vede che il legando è contenuto in una tasca formata
da due avvolgimenti che sporgono dalla catena C (rosa) e dalla parete
beta pieghe della colonna D (ciano).
Per studiare meglio il legando selezionate CCE-1206 e
salvatelo come CCE.pdb.
Cliccate col pulsante destro su 1uv6_tut e scegliete Delete
per cancellare tutto.
Aprite ora CCE.pdb (che
potete anche scaricare da qui).
Scegliete la rappresentazione Balls and Sticks e coloratela con
balls, sticks, By atom type.
Aggiungete gli idrogeni con Edit / Hydrogens / Add , OK.
Regolate il valore minimo di fog in Tools intorno a 11,
un poco sotto il valore massimo.
Otterrete l'immagine mostrata qui sotto (figura 5).
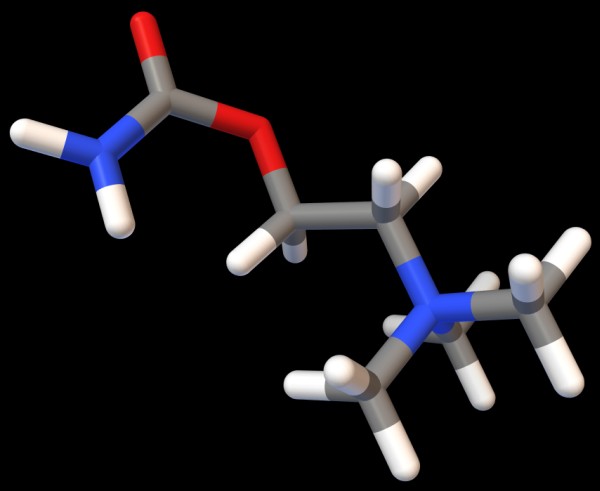
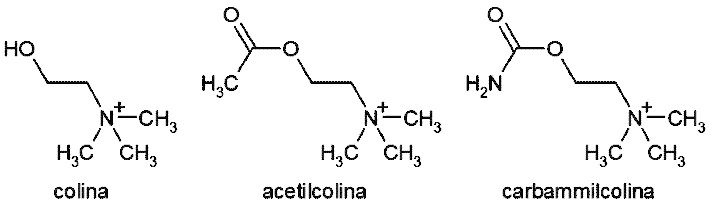
Come si vede qui sopra in figura 5b, la carbammilcolina è
leggermente più polare dell'acetilcolina (ha un NH2
al posto di un CH3)
e questo le consente di legarsi con più forza nel sito di legame
e di rimanere nella proteina anche durante la cristallizzazione.
Ricordiamo che queste strutture si ottengono dalla diffrazione di raggi
X su cristalli di proteine.
Per capire come il legando è alloggiato nella proteina, bisogna
individuare e studiare il sito di legame della proteina.
|
|
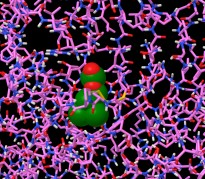
fig. 6
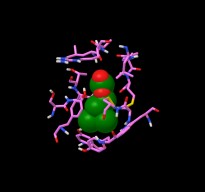
fig. 6a
figura 7
|
|
Individuare il sito di legame
Il sito di legame è costituito da tutti gli amminoacidi che circondano
il legando.
Chiudete e riaprite MGL Tools.
Aprite 1uv6_tut, selezionate CCE-1206 nella catena C ed
eliminatelo con Edit / Delete / Delete Selected Atoms.
Salvate la la proteina senza il legando con il nome 1uv6_pep.
Cancellate 1uv6_tut e aprite 1uv6_pep.
Aprite ora anche CCE.pdb e avrete proteina e legando in due molecole
distinte.
Aggiungete gli idrogeni: Edit / Hydrogens / Add.
Lasciate visibili solo gli idrogeni polari: Edit / Hydrogens / Merge
Non-Polar.
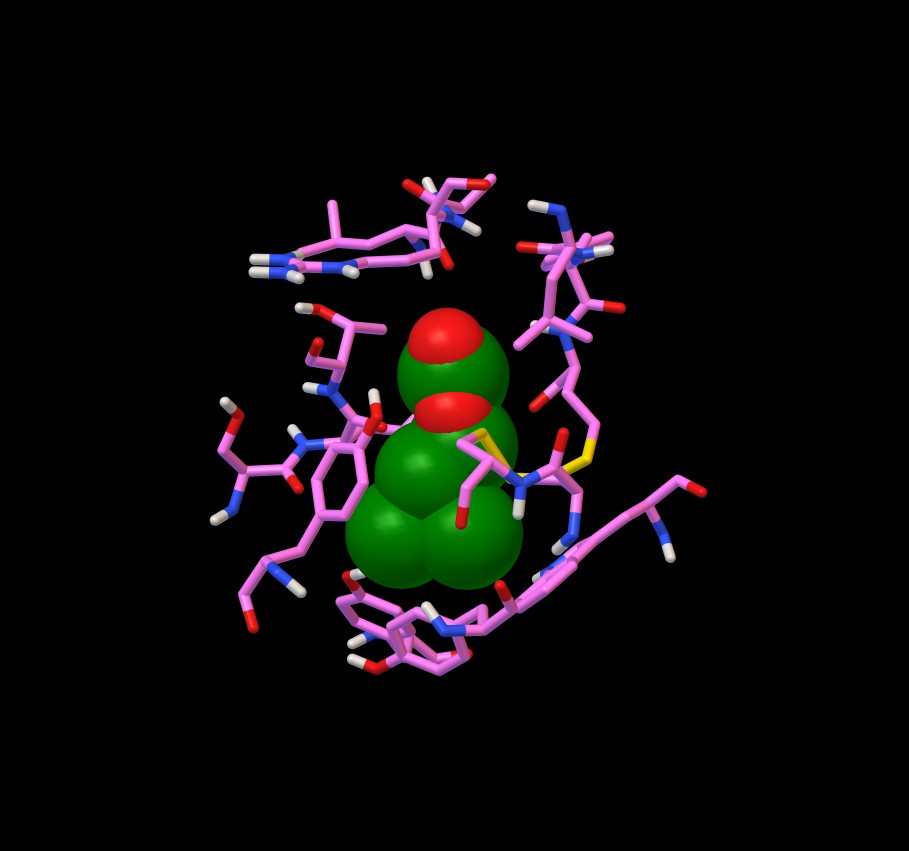
Rappresentando il legando con Sfere e la proteina con Balls
and Sticks si vede che il legando è circondato da una selva
di amminoacidi come è mostrato in figura 6.
Selezionate gli amminoacidi del sito di legame con questa procedura:
Selezionate CCE (colonna S)
Premete Select / Spherical Region e, nella finestra di dialogo,
aumentate il raggio a 5.5 A, cliccate poi current selection / select
/ close.
Quindi cliccate Select / Set Selection Level / Residue.
Vedrete gli amminoacidi attorno al legando selezionati con crocette
gialle.
Cliccate Select / Invert Selection per invertire la selezione
e Edit / Delete / Delete Selected Atoms per eliminare tutti gli
amminoacidi più lontani. Otterrete l'immagine di figura 6a.
Selezionate 1uv6_pep e salvate col nome 1uv6_sito,
dato che contiene solo gli amminoacidi del sito di legame: 8 amminoacidi
della catena C e 7 amminoacidi della catena D..
Potete anche continuare con gli amminoacidi che avete sullo schermo,
ma se avrete problemi potete ricaricare 1uv6_sito.
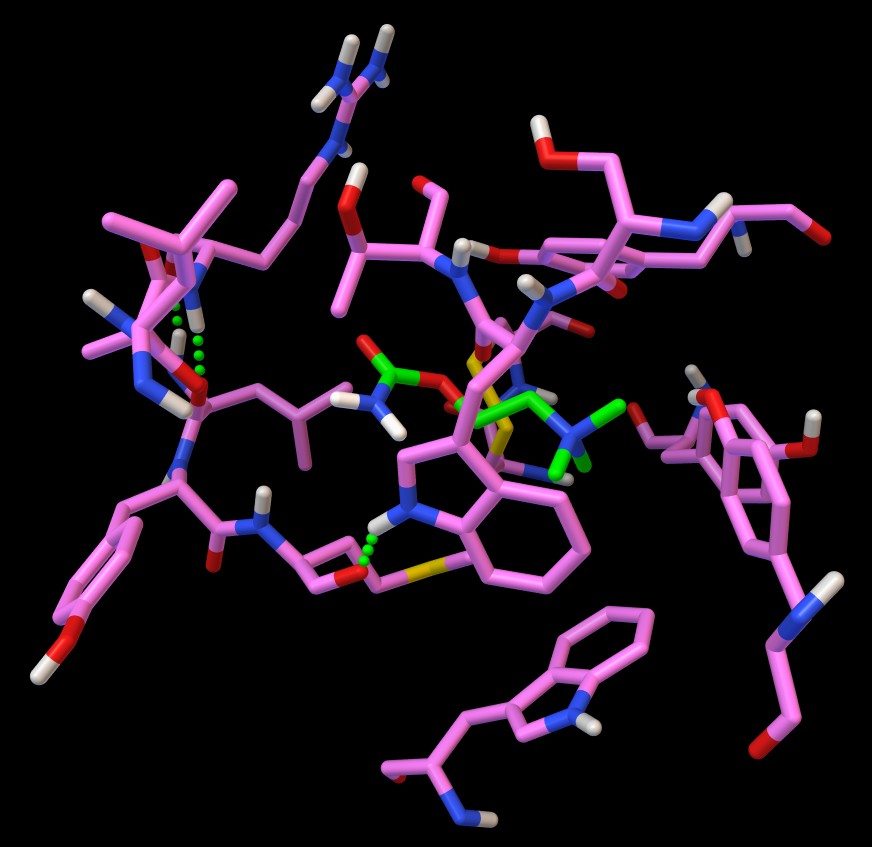
Cliccate Hydrogen Bonds / Build / Set Parms+Build, e si ottengono
tre legami idrogeno
quindi cliccate Hydrogen Bonds / Display / As Spheres.
Otterrete l'immagine qui sotto (figura 7).
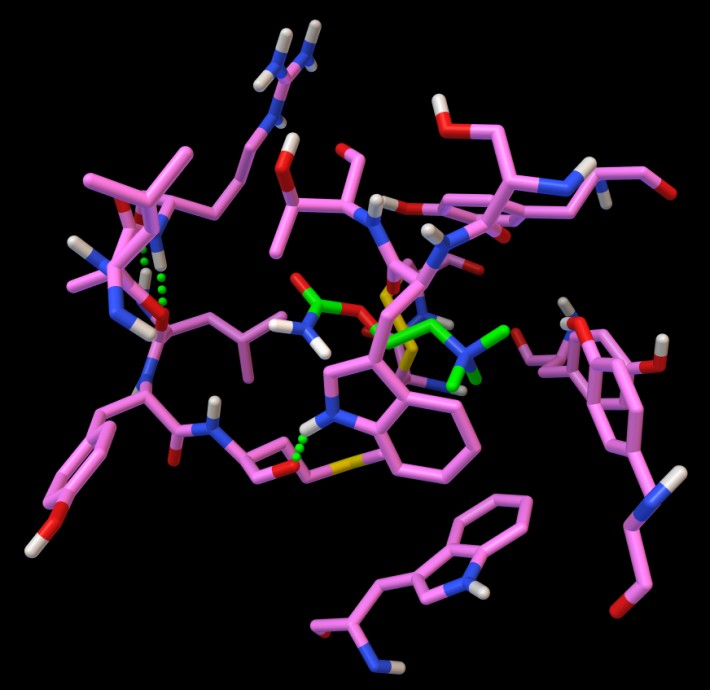
Il legando è stato rappresentato con Balls
and Sticks e reso più visibile colorandolo con l'opzione
balls, sticks, Carbon only, Custom color e poi scegliendo il
verde chiaro dalla tavolozza.
Osservate che la zona destra del legando, che contiene il gruppo ammonio
positivo RN+(CH3)3,
è circondata da tre tirosine, due sulla destra e una sopra,
(aromatiche, negative a pH neutro) e da due triptofani, in basso
e verso di noi, (aromatici, apolari).
Il lato sinistro del legando, che contiene un estere e un'ammide (polare)
è circondato da amminoacidi impegnati nei tre legami idrogeno,
che formano quindi una zona polare.
Le interazioni legando - sito di legame possono essere analizzate
meglio di così usando appositi programmi come AutoDock Vina.
Questi programmi calcolano l'energia dell'interazione legando-proteina
mentre cercano di posizionare il legando nel sito di legame nel miglior
modo possibile, un'operazione chiamata docking molecolare (docking
= mandare in porto).
I programmi di docking molecolare consentono anche di studiare l'interazione
con un legando diverso da quello presente nella proteina cristallizzata.
Nella prossima lezione faremo il docking di un farmaco antiinfiammatorio,
ibuprofene, nella sua proteina bersaglio, COX2.
Autore: prof Mauro Tonellato
|
|