|
Pianeta
Chimica.it
Responsabile:
Prof. Mauro Tonellato
|
|
MODELLISTICA
MOLECOLARE
Docking Molecolare
con AutoDock Vina
DOCKING
su COX2
|
|
|
| |
|
|
|
|
|
Chimica al computer con
MGL Tools / AutoDock
 Struttura delle Proteine
Struttura delle Proteine
Proteine e Legandi
Docking su COX2
Progettare nuovi farmaci
Chimica al computer con
ArgusLab
1-butene
e 2-butene
Carbocatione
1° e 2°
Acetone
e tautomeria
Dieni
coniugati (pdf)
Benzene
e aromaticità (pdf)
Legame covalente
Conformazione alcani
Ponte Cloronio
Diels-Alder
Enolo
Mono e Disostituito (pdf)
 Chimica al Computer
Chimica al Computer
PianetaChimica home
|
|
Docking Molecolare significa letteralmente mandare in porto
una molecola, si tratta di una simulazione al computer che calcola
la posizione più conveniente che un legando (una piccola
molecola organica) può assumere all'interno del sito attivo di
un enzima. Se il legando non è quello naturale dell'enzima, si
comporta come un inibitore competitivo e blocca temporaneamente
l'attività dell'enzima. Questa tecnica è utilizzata per
progettare al computer nuovi farmaci.
Uno dei programmi più usati per fare Docking
Molecolare è AutoDock
Vina (gratuito, open source, creato dal Dr. Oleg Trott del The
Scripps Research Institute) che ha raggiunto la velocità e l'affidabilità
di altri costosi programmi commerciali. Quello che segue è un
AutoDock Vina tutorial.
Per fare docking molecolare con AutoDock Vina non servono attrezzature
professionali, basta un normale PC e seguire la procedura mostrata qui
sotto.
Se siete insegnanti di chimica, potreste proporre ai vostri studenti
un'esperienza pratica di docking molecolare nel laboratorio di informatica
della scuola.
I software da scaricare sono i seguenti:
MGL Tools http://mgltools.scripps.edu/downloads
(prepara proteina e legando e valuta i risultati)
AutoDock Vina http://vina.scripps.edu/download.html
(esegue il docking molecolare)
Avogadro http://avogadro.cc/wiki/Main_Page
(crea nuove molecole da usare come legandi)

COX2, cicloossigenasi 2, è l'enzima
che trasforma l'acido arachidonico in prostaglandina, un mediatore del
dolore e dell'infiammazione. COX2 è il bersaglio dei FANS, farmaci
antiinfiammatori non steroidei, tra i quali, i più famosi sono
aspirina e ibuprofene.
Celecoxib è un farmaco antiinfiammatorio di nuova generazione.
In questo primo AutoDock Vina tutorial faremo
il docking di celecoxib all'interno dell'enzima COX2 (3LN1.pdb).
Confronteremo la posizione calcolata da AutoDock Vina con la
posizione reale che questo farmaco assume nell'enzima ricavata
dai dati di cristallografia a raggi X.
In questo modo potremo verificare l'accuratezza del docking molecolare
di AutoDock Vina.
Infine faremo il docking di ibuprofene nello stesso enzima COX2
per capire quale sia il farmaco migliore.
Nella prossima lezione progetteremo un nuovo farmaco antiinfiammatorio
e lo confronteremo con i farmaci commerciali.
I passaggi di questo primo AutoDock Vina tutorial sono:
-- Installazione del software
-- Preparazione dei file pdb della proteina e del legando
-- Preparazione dei file pdbqt della proteina e del legando
-- Individuazione del sito di legame e scrittura del file config.txt
-- Docking
-- Valutazione dei risultati
N.B. Cliccando sulle immagini qui sotto potrete vederle in versione
ingrandita.
|
|
|
figura 1

fig. 2
|
|
Installare il software
=> Scaricate i file indicati sopra e avrete i
tre file seguenti:
autodock_vina_1_1_2_win32.msi
mgltools_win32_1.5.6_Setup.exe
Avogadro-1.1.1-win32.exe
=> Installate i tre programmi.
=> Scaricate la directory COX2 che
contiene i file PDB per questa lezione.
=> La directory COX2 deve essere decompressa nel disco C in modo
da creare il percorso C:/COX2
=> Copiate il file Vina.exe nella directory di lavoro
(Programmi/The Scripps Research Institute/Vina.exe ===>
C:/COX2)
Preparare i file pdb della proteina
e del legando
Partendo dal file 3LN1.pdb (che contiene
l'enzima COX2 legato a celecoxib) dovete ottenere due
file distinti, uno contenente solo la proteina che chiamerete
3LN.pdb e l'altro contenente solo il legando che chiamerete
CEL.pdb.
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
1) Estraete
il legando celecoxib e salvatelo come CEL.pdb
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
> > > Lanciate AutoDockTools partendo
dall'icona sul desktop ---
> > > Impostate la directory COX2
come directory di default in AutoDockTools ---
Cliccate File => Preferences => Set
In Startup Directory scrivete => C:\COX2 premete Make
Default e Set => Dismiss
> > > Aprite la proteina 3LN1.pdb
---
Pulsante destro su All Molecules => Read Molecule
in C:/COX2 scegliete 3LN1.pdb => Apri
> > > Eliminate le catene B, C e D, lasciando
solo la catena A ---
Cliccate sul triangolo a fianco di 3LN1. Compaiono le
4 catene A, B, C, D. Selezionate le catene B, C, D cliccando il quadrato
a destra delle catene (colonna S, select).
Cliccate poi Edit => Delete => Delete Selected
Atoms come nella figura qui sotto (figura 1). Rispondete
CONTINUE e le catene selezionate saranno cancellate.
Ora cliccate sull'icona bersaglio (in alto sotto edit) per centrare
la catena A nello schermo.

> > > Eliminate le molecole d'acqua ---
Edit => Delete water => Continue
> > > Salvate il legando celecoxib (CEL)
contenuto nella catena A ---
Nella finestra di sinistra cliccate sul triangolo a fianco della
catena A. Si apre la sequenza dei residui contenuti in A
Scorrete fino alla fine degli amminoacidi e vedrete il residuo CEL
682. Cliccate sul quadrato a destra (colonna S, select)
e vedrete che gli atomi del legando si evidenziano in giallo come in
fig.2.
Cliccate File => Save => Write PDB
Scrivete al posto del nome del file corrente (3LN1) il nome del legando
(CEL)
c:\COX2\CEL.pdb => OK
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
2) Eliminate
il legando e salvate la proteina senza legando col nome 3LN.pdb
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
> > > Cancellate il legando ---
Mentre il legando è ancora selezionato (giallo) cliccate su
Edit => Delete => Delete Selected Atoms
=> Continue
> > > Salvate la proteina senza legando
col nome 3LN.pdb ---
Cliccate File => Save => Write PDB
Scrivete al posto del nome del file corrente (3LN1) il nome della proteina
con la sola catena A, senza legando e senz'acqua: 3LN.pdb
c:\COX2\3LN.pdb => OK
> > > Cancellate ora la proteina dallo
schermo ---
Pulsante destro su 3LN1 nella finestra di sinistra =>
Delete
|
|
|
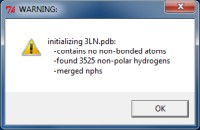
fig. 3
Fig.4

fig. 5 |
|
Preparare i file pdbqt della proteina e del legando
I file pdb contengono informazioni sulla posizione nello spazio
dei vari atomi di una molecola. Per realizzare il docking, AutoDock
Vina ha bisogno di informazioni supplementari come la carica
parziale, la posizione degli idrogeni polari, i legami che possono ruotare
nel legando, ecc.
Queste informazioni vengono scritte in un nuovo file con formato
pdbqt.
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
1) Proteina.
Aggiungete gli idrogeni e salvate proteina.pdbqt
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
> > > Aprite la proteina 3LN.pdb ---
Pulsante destro su All Molecules => Read Molecule
scegliete 3LN.pdb => Apri.
> > > Aggiungete gli atomi di idrogeno
---
Edit => Hydrogens => Add => All
=> OK
> > > Salvate la proteina in formato
pdbqt ---
Cliccate Grid => Macromolecule => Choose
=> 3LN => Select Molecule => OK
Compare una finestra (fig.3) che dice che sono stati trovati 3525 idrogeni
non polari e che sono stati nascosti (merged). Ora restano solo gli
idrogeni polari, quelli capaci di fare legami idrogeno.
=> OK (fig.4)
Il programma chiede di salvare la proteina così modificata come
3LN.pdbqt => OK
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
2) Legando.
Aggiungete gli idrogeni, determinate il numero di torsioni e salvate
legando.pdbqt
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
> > > Cancellate la proteina ---
Pulsante destro su 3LN => Delete
> > > Aprite il legando CEL.pdb ---
Pulsante destro su All Molecules => Read Molecule
scegliete CEL.pdb => Apri.
> > > Aggiungete
gli atomi di idrogeno a CEL ---
Edit => Hydrogens => Add => OK
> > > Scegliete
CEL come legando ---
Cliccate Ligand => Input => Choose =>
CEL => Select Molecule for AutoDock 4 => OK
Il programma ha aggiunto le cariche parziali agli atomi in base all'elettronegatività,
ha individuato 12 idrogeni non polari e li ha nascosti lasciando visibili
solo quelli polari, 15 carboni aromatici, 5 legami ruotabili e ne ha
scelti 5 (fig.5)
> > > Salvate
il legando in formato pdbqt --
Ligand => Output => Save as PDBQT => CEL.pdbqt
=> Salva
> > > Cancellate
CEL ---
Pulsante destro su CEL => Delete e poi su 3LN
=> Delete
|
|
|
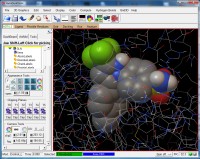
fig. 6
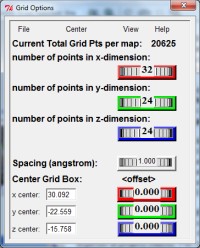
fig. 7
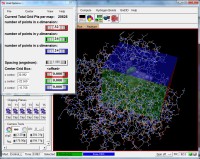
fig. 8

fig. 9
|
|
Individuare il sito di legame e scrivere il file config.txt
La zona della proteina dentro alla quale AutoDock Vina cerca di fare
il docking è delimitata da un cubo colorato chiamato GridBox.
Il sito di legame deve essere tutto contenuto all'interno della
GridBox.
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
1) Determinate
posizione e dimensioni della GridBox
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
> > > Aprite
la proteina 3LN.pdbqt e il legando CEL.pdbqt---
Pulsante destro su All Molecules => Read Molecule
=> 3LN.pdbqt => Apri
Pulsante destro su All Molecules => Read Molecule
=> CEL.pdbqt => Apri.
> > > Scegliete la macromolecola e il
legando ---
Grid => Macromolecule => Choose => 3LN
=> Select Molecule => OK
(rispondete yes alla richiesta di conservare le cariche parziali
già calcolate per la molecola)
Grid => Set Map Types => Ligand => Choose
ligand => CEL => Select ligand
> > > Rendete
il legando ben visibile per centrare più facilmente la GridBox
su di lui ---
A fianco della proteina 3LN lasciate selezionato il cerchietto
L (lines)
A fianco del legando CEL deselezionate il cerchietto L
e scegliete il cerchietto C (atomic spheres)
Nel triangolo Cl (color) scegliete => cpk => By
atom type
Se le immagini sono poco brillanti andate nella schermata Tools
(nella finestra a sinistra) e impostate i valori iniziali di fog
a 142 (fig.6)
La GridBox deve essere centrata sul sito di legame, in
generale questo va fatto spostandola manualmente con le tre rotelle
rossa, verde e blu fino a quando il suo centro (crocetta gialla) è
nel centro del sito di legame.
In questo caso il sito di legame dell'enzima è facilmente individuabile
dato che 3LN.pdb conteneva un legando al proprio interno.
Per centrare la GridBox sul sito di legame di 3LN è sufficiente
centrare la GridBox sul legando CEL.
>>> Aprite la finestra di dialogo per
generare la GridBox ---
Grid => GridBox
>>> Regolate l'unità di misura
su 1 Angstrom ---
Spacing (angstrom) => 1.00
>>> Riducete le dimensioni della GridBox
---
number of points in x, y, z dimension => 20, 20, 20
>>> Centrate la GridBox sul legando ---
Center => Center on ligand
Vedrete un cubo colorato centrato sul legando con dimensioni adattate
al legando (32, 20, 24)
in questo caso è preferibile allargare un po' la GridBox
fino a 32, 24, 24 angstrom. (fig.7)
>>> Annotate le dimensioni della GridBox
---
size_x = 32
size_y = 24
size_z = 24
>>> Annotate i valori
del Centro della GridBox approssimati ad un solo decimale
---
In questo caso i valori sono
center_x = 30.1
center_y = -22.6
center_z = -15.8
>>> Salvate la GridBox ---
File => Close saving current (fig.8)
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
2) Scrivere il file config.txt
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
AutoDock Vina cerca nel file config.txt le informazioni minime
che riguardano il docking.
receptor = . . . nome della proteina
ligand = . . . nome del legando
center_x = . . . coordinate del centro della GridBox
center_y = . . .
center_z = . . .
size_x = . . . dimensioni della GridBox in Angstrom
size_y = . . .
size_z = . . .
out = . . . nome del file in cui AutoDock Vina pone i
risultati del docking
In questo caso, dunque, il file config.txt va scritto così:
receptor = 3LN.pdbqt
ligand = CEL.pdbqt
center_x = 30.1
center_y = -22.6
center_z = -15.8
size_x = 32
size_y = 24
size_z = 24
out = 3LN_CEL_vina.pdbqt
> > > Usate copia-incolla per scrivere queste righe
in Notepad o in un qualsiasi editor di testo e salvate
il file col nome config_3LN_CEL.txt nella directory di lavoro
C:/COX2 (fig.9) < < < < < < < < <
|
|
|

fig. 10

fig. 11
|
|
Docking
Per fare il docking con AutoDock Vina,
la directory di lavoro deve contenere i seguenti 4 file:
proteina.pdbqt
legando.pdbqt
config.txt
vina.exe
--- Verificate che in C:/COX2 siano presenti i seguenti file ---
3LN.pdbqt
CEL.pdbqt
config_3LN_CEL.txt
vina.exe
> > > Lanciate il docking ---
Cliccate Run => Run AutoDock Vina
Si apre una finestra di dialogo.
in Vina Program Pathname cliccate => Browse => vina.exe
=> Apri
In Config Filename cliccate => Browse => config_3LN_CEL.txt
=> Apri => Launch (fig.10)
Il processo di docking può essere più o meno lungo a seconda
della potenza di calcolo del computer in uso. Con un buon computer multiprocessore
questo docking richiede circa 25 secondi.
Durante il docking resta aperta una finestra di dialogo (fig.11).
Se questa finestra si apre e si chiude subito, allora avete compiuto qualche
errore. Verificate per prima cosa la correttezza dei nomi dei file
scritti in config_3LN_CEL.txt. Ricordate che i nomi dei file e delle directory
non devono mai contenere spazi.
|
|
|
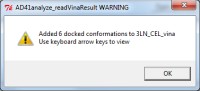
fig. 12
fig.13
|
|
Valutare i risultati
Per valutare la bontà del docking, bisogna caricare le molecole
del legando nella posizione calcolata da AutoDockVina che vengono dette
pose. Il programma dà a ciascuna posa un punteggio che esprime
l'intensità (in kcal/mol) dell'interazione legando-proteina.
Poi è utile osservare il legando all'interno della tasca del
sito di legame per valutare se la riempie completamente, infine è
opportuno osservare il legando circondato dagli amminoacidi della tasca,
per vedere meglio l'interazione di ogni amminoacido col legando.
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
1) Osservate le pose
migliori in confronto con il legando originale
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
> > >
Caricate il file che contiene i risultati del Docking ---
Analize => Dockings => Open Autodock Vina Results
=> 3LN_CEL_vina.pdbqt => Apri
=> Single molecule whith multiple conformations => OK
(fig.12).
Il programma ha caricato una nuova molecola 3LN_CEL_vina che
contiene 6 diverse conformazioni calcolate da AutoDock Vina, e consente
di vederle in sequenza dalla migliore (-12.5 kcal/mol) alla peggiore
(-9.7 kcal/mol) premendo freccia a destra o a sinistra.
> > >
Osservate le pose calcolate da AutoDock Vina in confronto al legando
originale ---
Su 3LN spegnete il cerchietto L per disattivare
la vista a linee. La proteina sparisce dallo schermo.
Su CEL accendete solo il cerchietto B per vederlo
con Sticks and Balls.
Inoltre, nella colonna Cl cliccate Balls, Sticks,
Carbon only => By residue type (oppure verde)
CELL avrà i carboni verde chiaro
Su 3LN_CEL_vina accendete solo il cerchietto B per vederlo
con Sticks and Balls.
Inoltre, nella colonna Cl cliccate Balls, Sticks,
Carbon only => Custom color (arancione)
3LN_CEL_vina avrà i carboni arancione. Se lo vedete un po' scuro,
alzate il valore di inizio di fog nella finestra Tools.
Ora vedrete nello schermo il legando originale CEL (verde) e il legando
posizionato da Vina (arancione) con l'indicazione dell'energia di interazione
proteina-legando, in questo caso -12,5 kcal/mol. (fig.13).
Questa è la migliore delle sei pose trovate. Per vedere le altre
usate la freccia destra e sinistra, se l'immagine sparisce riaccendete
il cerchietto B.
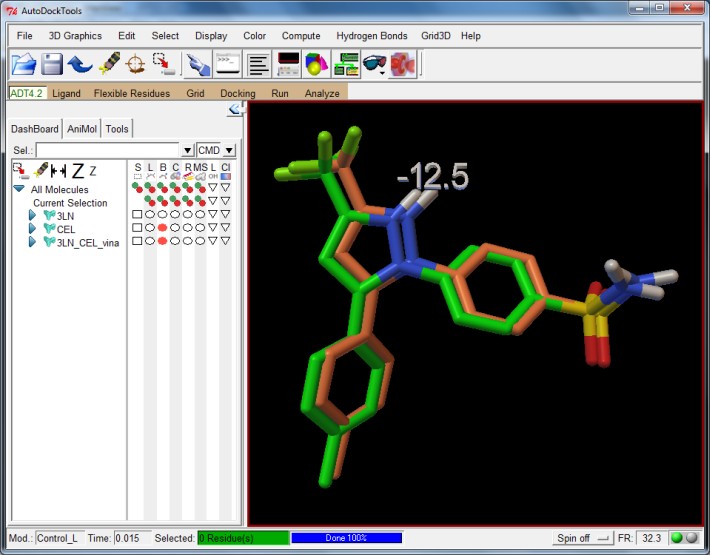
La posa migliore calcolata da AutoDock Vina
(arancione) si sovrappone in modo quasi perfetto al legando originale
(verde) tranne che per un minimo slittamento verso destra.
|
|
|
|
|
|
|
|
|
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
2) Osservare il legando all'interno della tasca del sito attivo
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Cancellate il legando originale CEL.
Create la superficie molecolare della proteina (cerchietto MS).
Colorate la superficie secondo la polarità: triangolino Cl,
cliccate su MSMS-MOL e poi cliccate su By polarity (fig.14)
Per otterrete l'immagine mostrata qui sotto cliccate su Tools
e poi cliccate su F (front) e sul terzo riquadro (poligoni ombreggiati),
poi su B (back) e sul secondo riquadro (poligoni a rete) (fig.15).
Regolate ClipZ (in basso nella finestra Tools) tra 19 e 32 per
togliere dalla vista le porzioni della superficie anteriori e posteriori
alla tasca.
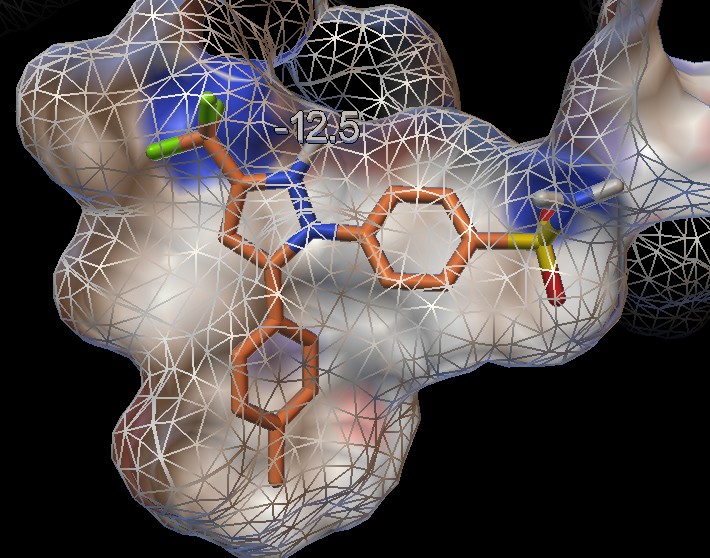
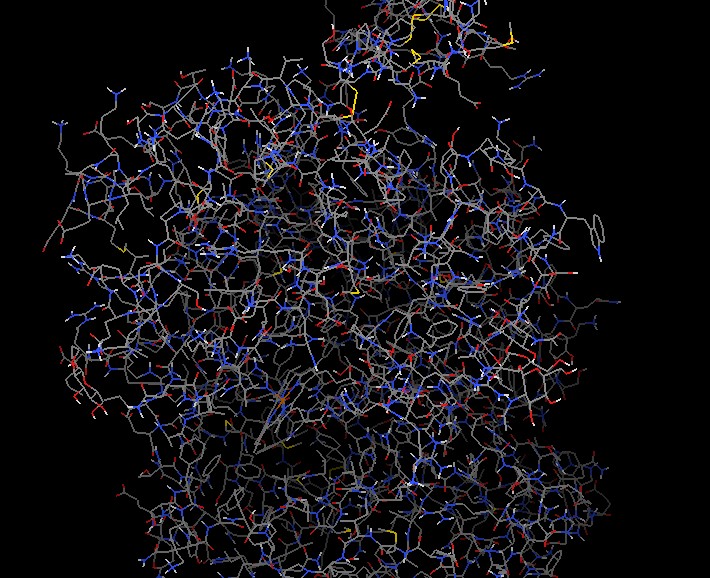
Otterrete la struttura mostrata qui sopra (fig.16) nella quale si può
vedere molto bene che il farmaco celecoxib riempie la tasca dell'enzima
in modo ottimale.
In alto il gruppo CF3 si colloca in
una zona apolare a sinistra (bianca), ma basica sul fondo (blu).
A destra la solfonammide SO2NH2
si avvicina con gli ossigeni ad una zona leggermente basica sul fondo
(blu), ma verso di noi avvicina il gruppo NH2 ad una zona leggermente
acida (rossa) non visibile da questa inquadratura.
In basso il gruppo CH3 entra in una
zona che è sostanzialmente apolare (bianca), ma ha zone leggermente
acide (rosse) ai lati.
(Ricordiamoci di questo particolare quando progetteremo una variante
più efficace di questo farmaco nella prossima lezione!).
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
3) Creare un'animazione
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Nel menù 3D Graphics => Spin Bounce Oscillate
=> Rock angle 60 => Bounce
Nel menù 3D Graphics => Video Recorder; usate i tasti
per iniziare e fermare la registrazione
Dovreste ottenere un'animazione simile a questa COX2-CEL_rete.mpg
Un altro modo di valutare l'interazione legando-proteina è osservare
il legando e gli amminoacidi che lo circondano nel sito di legame. In
questo modo si possono valutare meglio le interazioni polari o apolari
con i singoli amminoacidi.
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
4) Osservare il legando
circondato dagli amminoacidi del sito attivo
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
> > > Cancellate ogni molecola presente
---
> > > Caricate la posa migliore
---
Analize => Dockings => Open Autodock Vina Results
=> 3LN_CEL_vina.pdbqt
> > > Caricate la proteina 3LN.pdbqt
---
> > > Rappresentate il legando con sfere
--- (cerchietto C)
> > > Aggiustate il valore iniziale di
fog --- (in Tools) otterrete l'immagine qui sotto (fig.17)
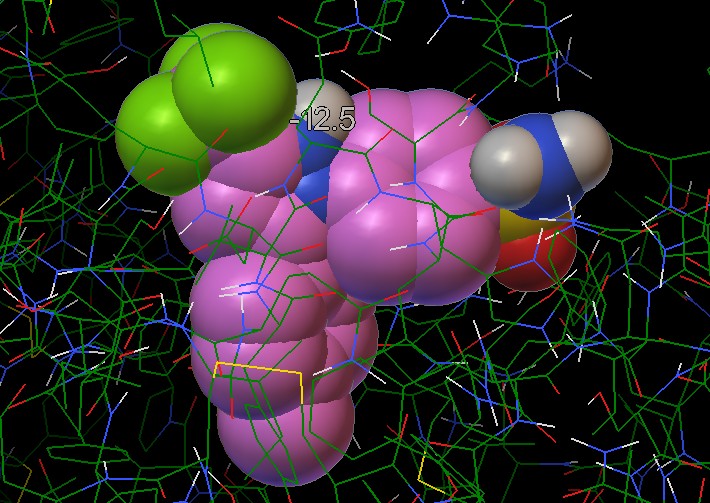
Dato che l'immagine è troppo confusa, eliminate dalla proteina
tutti gli amminoacidi ad eccezione di quelli che sfiorano il legando.
> > > Selezionate il legando ---
(cerchietto S)
> > > Selezionate gli amminoacidi vicini
al legando ---
Select => Select spherical region => 5 angstrom
=> Center spherical region on current selection
Cliccate su select , close. (fig.18)
Select => Set selection level > Residue (sono
selezionati legando e AA vicini)
Select => Invert selection => all => OK
(è selezionato tutto fuorchè legando e AA vicini)
Edit => Delete => Delete selected atoms =>
Continue (restano solo legando e AA vicini)
> > > Salvate gli AA vicini col nome
AA ---
Selezionate gli amminoacidi (quadratino S a fianco di 3LN) =>
File => Save => Write PDB => Cambiate
il nome della selezione scrivendo AA al posto di 3LN => OK
Cancellate ora 3LN e caricate AA
Rappresentate gli amminoacidi con Sticks and Balls (cerchietto B)
Colorateli in modo che siano ben riconoscibili
Triangolino Cl => All rappresentations => Carbon
only => by residues type (Shapely)
Otterrete l'immagine qui sotto (fig.19-a)
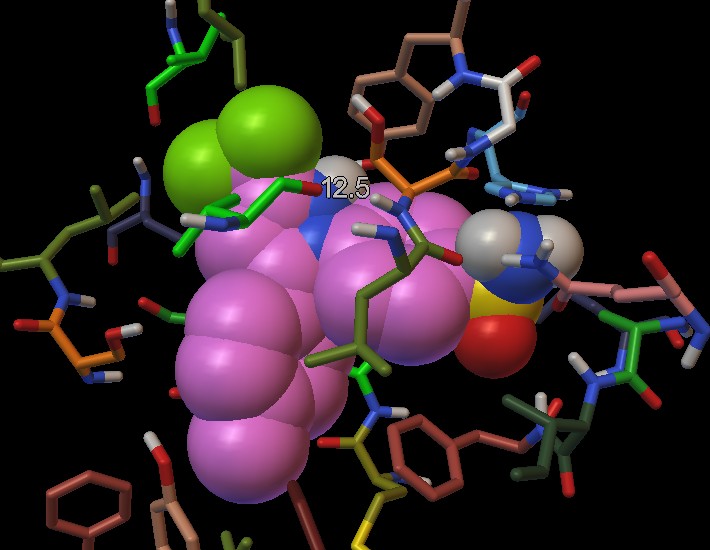
Questa immagine ci fornisce informazioni solo su un lato della tasca,
per questo è utile osservare anche il lato opposto (fig.19-b).

In alto, vicino al gruppo CF3, si vedono due
leucine (verdi scure, apolari) e una valina (verde chiaro, apolare),
ma sul davanti in questa seconda immagine si vede una arginina (blu)
e una tirosina (beige), molto più polari (vedremo più
avanti che con questi due amminoacidi interagisce il carbossile dell'ibuprofene).
Vicino agli ossigeni del gruppo SO2NH2,
sulla sinistra di fig.19-b, si vede una arginina (blu), e una istidina
(azzurra) mentre vicino all'NH2 dello stesso gruppo, sulla destra di
fig.19-a, si vedono gli ossigeni del carbossile di una serina (arancione),
di una leucina (verde) e di una glutammina (rosa).
In basso, vicino al gruppo CH3, ci sono gruppi
apolari: una metionina (gialla), una fenilalanina (marrone), una leucina
(verde scuro), un triptofano (marrone). Vicino al CH3,
nella figura 19-a sulla sinistra, si vedono, però, anche gruppi
polari: una tirosina (beige) e una seina (arancione).
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
5) Creare un'animazione
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Nel menù 3D Graphics => Spin Bounce Oscillate
=> Rock angle 120 => Bounce
Nel menù 3D Graphics => Video Recorder; usate i tasti
per iniziare e fermare la registrazione
Dovreste ottenere un'animazione simile a questa COX2-CEL_AA
|
|
|
|
|
|
Docking con ibuprofene
Per eseguire il docking con un altro farmaco antiinfiammatorio, la procedura
è più breve perchè 3LN.pdbqt, GridBox, config.txt
sono già pronti, basta preparare il legando e adattare il file
config.txt ai nuovi dati.
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
1)
Preparare il file legando.pdbqt
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Cancellate ogni molecola dal programma
Aprite il file dell'ibuprofene IBP.pdb che si trova nella directory
di lavoro COX2
Aggiungete gli idrogeni (Edit => Hydrogens => Add => All)
Trasformate IBP in un legando (Ligand => Input => Choose =>
IBP => Select Molecule for AD4)
Salvate IBP.pdbqt (Ligand => Output => Save as PDBQT => Salva)
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
2)
Preparare il file config.txt
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Usando Notepad sostituite IBP al posto di CEL nel vecchio config.txt
preparato prima, poi salvate il file con il nome config_3LN_IBP.txt
receptor = 3LN.pdbqt
ligand = IBP.pdbqt
center_x = 30.1
center_y = -22.6
center_z = -15.8
size_x = 32
size_y = 24
size_z = 24
out = 3LN_IBP_vina.pdbqt
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
3)
Lanciate il nuovo docking
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Run => Run AutoDock Vina => individuate i file Vina.exe
e config_3LN_IBP_txt => Launch
Il docking termina in circa 8 secondi
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
4)
Valutare i risultati del docking
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Analize => Dockings => Open Autodock Vina Results
=> 3LN_IBP_vina.pdbqt => Apri
=> Single molecule whith multiple conformations => OK
Cancellate il legando IBP
Aprite il legando CEL, cioè il celecoxib originalmente presente
nel sito di legame
Rappresentate entrambi i legandi con Sticks and Balls
Colorate 3LN_IBP di rosa (balls, sticks, carbons only, custom
color, rosa)
Colorate CEL di verde (balls, sticks, carbons only, custom color,
verde)
Qui sotto (fig.20) si vede la posa dell'ibuprofene (rosa) sovrapposta
a quella del celecoxib (verde)
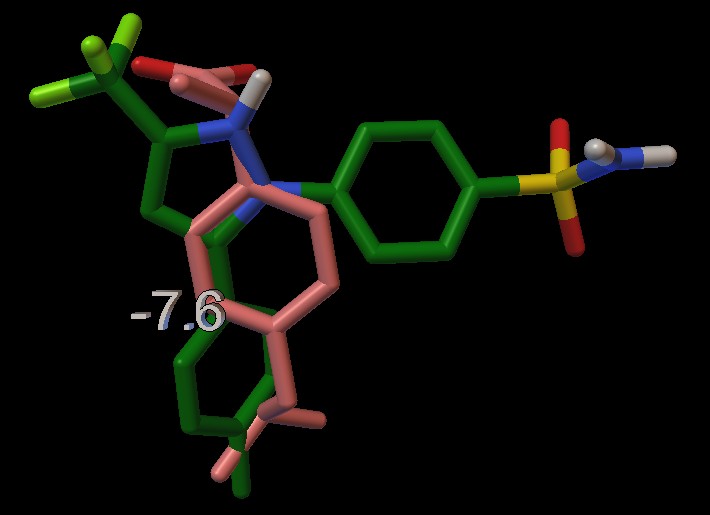
Notate il valore dell'interazione: -7,6 kcal/mol, molto più basso
di quello del celecoxib.
Questo valore va interpretato con molta prudenza perchè ci dà
un'idea solo dello stato finale del processo di interazione proteina-legando,
ma non ci racconta l'energia di desolvatazione, nè tiene conto
dell'entropia, inoltre è logico che una molecola più grande
dia una interazione maggiore perchè è maggiore la superficie
di contatto, ma una molecola più grande ha anche maggiori interazioni
col solvente prima di entrare nel sito di legame.
In ogni caso si nota che ibuprofene occupa una zona più limitata
della tasca enzimatica, mentre celecoxib si estende anche nella zona
a destra. Questo significa che celecoxib è un farmaco più
selettivo perchè ha bisogno esattamente di una tasca di questa
forma e dimensione per legarsi, mentre ibuprofene, essendo una molecola
più piccola, può entrare sia in tasche così grandi,
ma anche in quelle più piccole dalle quali celecoxib è
escluso.
Per esempio, l'enzima cicloossigenasi 1 (COX1) ha una tasca più
piccola di COX2 e ibuprofene può legarsi ad entrambi gli enzimi,
mentre celecoxib è specifico per COX2.
Bloccare l'azione delle COX2 significa fermare il dolore e l'infiammazione
che sono mediati dalle prostaglandine che questo enzima produce.
Bloccare l'azione delle COX1 significa impedire l'aggregazione piastrinica
e il controllo della produzione di HCl nello stomaco, che sono mediati
dalle prostaglandine prodotte da COX1.
Sono proprio questi i principali effetti collaterali dei farmaci antiinfiammatori
che colpiscono non solo le COX2, ma anche le COX1.
La rappresentazione dei due legandi all'interno della tasca di legame
dell'enzima COX2 ci può raccontare molto di più (fig.21).
Notiamo in basso che ibuprofene colloca i due gruppi CH3
del gruppo isobutilico dove celecoxib ne mette uno solo.

Create un animazione basata su quet'immagine
Nel menù 3D Graphics => Spin Bounce Oscillate
=> Rock angle 60 => Bounce
Nel menù 3D Graphics => Video Recorder; usate i tasti
per iniziare e fermare la registrazione
Dovreste ottenere un'animazione simile a questa COX2-IBP-CEL
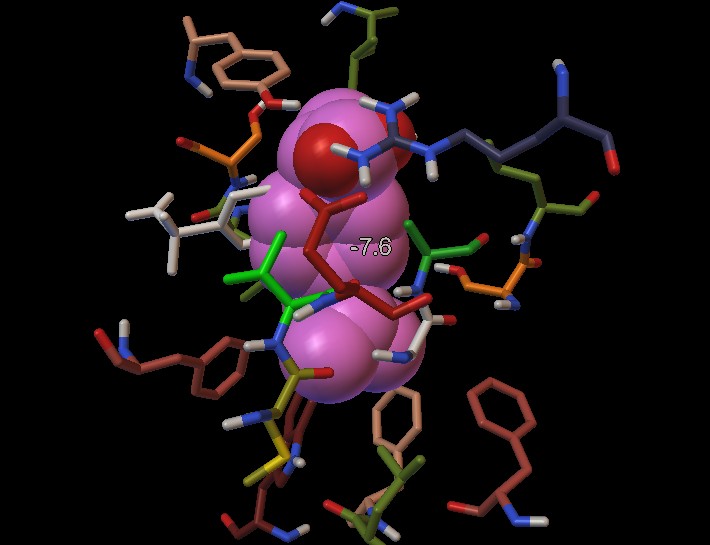
Infine nell'immagine qui sotto (fig.22)
vediamo l'ibuprofene circondato dagli amminoacidi che lo sfiorano nella
tasca di legame di COX2. L'immagine è leggermente ruotata per
mettere in evidenza l'interazione del carbossile di ibuprofene, in alto,
con due dei tre gruppi amminici (blu) dell'arginina sul davanti e, in
minor misura, con il gruppo OH (rosso) della tirosina (beige).
Notate, sulla sinistra, la struttura completamente bianca che sporge
dall'ibuprofene: è il celecoxib che riempie anche quel settore
della tasca di legame.
Nella prossima lezione impareremo a progettare nuovi legandi e, sottoponendoli
al docking molecolare, impareremo a valutare la loro capacità
di interagire con l'enzima. Potrete sfidare le grandi case farmaceutiche
scoprendo nuovi farmaci per combattere qualcuna delle malattie che ancora
oggi non hanno una cura.
Autore: prof Mauro Tonellato
|
|
|
|
|
|
Chimica al Computer
PianetaChimica home
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|