|
Pianeta
Chimica.it
Responsabile:
Prof. Mauro Tonellato
|
|
MODELLISTICA
MOLECOLARE CON MGL TOOLS
STRUTTURA DELLE PROTEINE
|
|
| |
|
|
|
|
Chimica al computer con
MGL Tools / AutoDock
 Struttura delle Proteine
Struttura delle Proteine
Proteine e Legandi
Docking su COX2
Progettare
nuovi farmaci
Chimica al computer con
ArgusLab
1-butene
e 2-butene
Carbocatione
1° e 2°
Acetone
e tautomeria
Dieni
coniugati (pdf)
Benzene
e aromaticità (pdf)
Legame covalente
Conformazione alcani
Ponte Cloronio
Diels-Alder
Enolo
Mono e Disostituito (pdf)
 Chimica al Computer
Chimica al Computer
PianetaChimica home
|
|
Le proteine sono macromolecole molto complesse, la loro struttura può
essere capita più facilmente con l'aiuto di un programma di modellistica
molecolare come MGL Tools. Questo programma ci mostra la proteina in
modo tridimensionale nello schermo del computer e ci consente di esaminarla
e manipolarla come se fosse un oggetto nelle nostre mani. Questo non
solo ci aiuta a capire meglio il significato di struttura primaria,
secondaria, terziaria e quaternaria delle proteine, ma ci permette di
comprendere meglio anche altre proprietà delle proteine, come
l'interazione legando - sito attivo negli enzimi o l'azione degli inibitori.
Questa lezione può essere affrontata in due modi.
1) Online. Potete leggere le istruzioni ed eseguirle passo passo
al computer con MGL Tools, aiutandovi con le illustrazioni che chiariscono
ogni passaggio.
2) In aula informatica con la classe. Se siete insegnanti di
chimica, potete adattare la lezione alle esigenze della vostra classe
e proporla in aula informatica ai vostri allievi.
La durata della lezione è di circa tre ore.
N.B. Cliccando sulle figure potrete vedere le immagini a pieno
schermo.

Gli argomenti di questa lezione sono:
-- Struttura primaria, secondaria, terziaria, quaternaria delle
proteine
|
|
|
Recettore dell'acetilcolina
In questa prima lezione sulle proteine, studieremo
il recettore dell'acetilcolina. Questo è un recettore proteico
che si trova sulla superficie delle cellule muscolari nelle sinapsi nervo-muscolo.
Quando l'impulso nervoso arriva alla fine del neurone, questo rilascia
un getto di neurotrasmettitori come l'acetilcolina, che giunge alla cellula
muscolare dove si lega a specifici recettori il cui poro centrale si apre
e lascia entrare un flusso di ioni sodio che innesca la contrazione muscolare.
Negli archivi PDB (Protein Data Bank) vi sono molte strutture di
recettori dell'acetilcolina. Qui useremo la struttura 1uv6 (mostrata
in figura 1), anzi, per cominciare, ne useremo solo un frammento creato
per questa lezione, 1uv6_tut (figura 2), che contiene solo 2 delle
5 catene proteiche che la molecola possiede in natura.
Aprire
il file PDB
Scaricate il file 1uv6_tut.pdb
e collocatelo in una cartella che chiamerete Acetilcolina nella
root directory c:\.
Lanciate MGL Tools e cliccate File / Preferences / Set per aprire
una finestra di dialogo, quindi scrivete in Startup Directory la
posizione della cartella Acetilcolina, c:\Acetilcolina.
In MGL Tools cliccate File / Read Molecule e aprite il file 1uv6_tut.pdb.
|
|

1uv6 - fig. 1

1uv6_tut - fig. 2 |
|
|
figura 3

fig. 4

fig. 5
figura 5b
|
|
Stuttura Primaria
La struttura primaria di una catena proteica è semplicemente
la sua sequenza di amminoacidi.
Per metterla in evidenza procedete così:
nella finestra di sinistra di MGL Tools, dove si trova la struttura
ad albero della molecola, cliccate sul triangolino azzurro a lato di
1uv6_tut e vedrete le subunità C e D presenti in questa
struttura.
Cliccate sul triangolino accanto a C vedrete la sequenza di amminoacidi
della catena C (figura 3).

Gli amminoacidi vengono numerati a partire dal lato N terminale.
La catena C inizia sul lato N terminale con LEU-1 e ASP-2 e termina
sul lato C terminale con LYS-204 e GLY-205 . Scorrendo verso il basso
gli amminoacidi della catena C troviamo, dopo GLY-205, la molecola del
legando contenuto in questa proteina, CCE-1206, carbammilcolina,
un analogo dell'acetilcolina.
Muovendo il mouse col pulsante sinistro premuto, si può
ruotare la proteina.
Agendo sulla rotellina centrale del mouse, si può ingrandire
o rimpicciolire l'immagine.
Muovendo il mouse col pulsante destro premuto, si può
traslare la proteina.
Gli amminoacidi che costituiscono la molecola sono rappresentati con
semplici linee. E' possibile usare rappresentazioni diverse usando
il menu della finestra di sinistra di MGL Tools (fig. 3).
Questo menù è composto da più colonne
Colonna S (Select) seleziona gli amminoacidi.
Colonna L (Lines) rappresenta gli amminoacidi con linee.
Colonna B (Balls and Sticks) rappresenta gli amminoacidi con
palline e bastoncini.
Colonna C (Spheres) rappresenta gli atomi con grosse sfere.
Colonna R (Ribbons) rappresenta la proteina con un nastro evidenziando
la sua struttura secondaria ad alfa elica, beta a pieghe o random coil.
Colonna MS (Molecular Surface) rappresenta la superficie della
proteina.
Colonna L (Labels) inserisce i nomi di atomi, molecole, amminoacidi
o catene.
Colonna Cl (Colors) modifica i colori di atomi, molecole, amminoacidi
o catene.
Proviamo a rappresentare i primi due amminoacidi con Balls and Sticks
con i colori tipici dei singoli atomi (C grigio, N blu, O rosso, S giallo,
H bianco).
Selezionate i primi due amminoacidi, LEU-1 (leucina) e ASP-2
(acido aspartico) cliccando sui due quadratini corrispondenti nella
colonna S (diventano gialli).
Cliccate sui cerchietti (diventano rossi) della colonna B di
LEU-1 e ASP-2 per rappresentarli con Balls and Sticks
Ruotate, traslate e ingrandite la proteina per portare in primo
piano gli amminoacidi selezionati.
Cliccate ora sul triangolino della colonna Cl per accedere al
menù di colorazione di LEU-1. Su questo cliccate balls,
sticks, e poi scegliete By atom type come mostrato in
figura 4.
Ripetete la procedura per ASP-2.
Deselezionate i due amminoacidi, LEU-1 e ASP-2
Otterrete l'immagine mostrata in figura 5 (cliccate sull'immagine
per ingrandirla).
Essendo LEU-1 l'amminoacido che inizia la catena, il suo gruppo alfa-amminico
è libero (atomo blu in alto), mentre il suo carbonio del carbossile
(carbonio grigio legato all'ossigeno rosso) è legato all'azoto
(blu) di ASP-2. Dal carbossile di ASP-2 si vede la catena proteica che
continua come linea sottile.
I due amminoacidi si riconoscono dalla loro catena laterale: una catena
di carboni ramificata, un isobutile, per la leucina 1. Una catenella
di due carboni che termina con un carbossile per l'acido aspartico 2.
Esercitatevi a riconoscere gli amminoacidi dalla loro catena laterale.
Sulla destra in figura si vede la catena laterale di un'asparagina,
sulla sinistra si notano le catene di un acido aspartico e di un'altra
asparagina.
Notate inoltre che il legame peptidico è planare, cioè
i 6 atomi legati agli orbitali sp2
del legame peptidico sono tutti sullo stesso piano.
Per evidenziare questo fatto selezionate i primi tre amminoacidi della
catena C (LEU1, ASP2, ARG3) e salvateli col nome 1uv6_aa
(che potete anche scaricare qui).
Cancellate la proteina 1uv6_tut e aprite il tripeptide appena salvato
1uv6_aa.
Selezionate la rappresentazione Balls and Sticks (colonna B),
colorate la struttura (colonna Cl) scegliendo (in Geometry) balls
e sticks e poi (in Color Scheme) By atom type (figura
4).
Nella finestra Tools impostate il valore minimo di fog
intorno a 26 (un po' minore di quello massimo, 35) per vedere ben illuminata
tutta la struttura.
Aggiungete gli idrogeni alla struttura, dato che mancano nel
file PDB originale.
Selezionate: Edit / Hydrogens / Add e poi cliccate OK.
Ora scegliete: Edit / Hydrogens / Merge non Polar e cliccate
Continue.
Nella struttura rimangono visibili solo gli idrogeni polari come si
vede in figura 5b.

I 6 atomi coplanari del primo legame peptidico
(tra Leu-1 e Asp-2) sono stati indicati con i numeri da 1 a 6.
Anche i successivi 6 atomi della catena sono coplanari (atomi 6, 7,
ecc) perchè sono coinvolti nel secondo legame peptidico (tra
Asp-2 e Arg-3), ma il loro piano è ruotato rispetto al
precedente.
Lo snodo tra i due piani è il carbonio alfa dell'acido
aspartico (atomo 6) che consente alla struttura di ruotare sia attorno
al legame 4-6 che attorno al legame successivo 6-7 dove 7 è il
carbonio del carbonile di Asp.
|
|

fig. 6
figura 7
|
|
Struttura Secondaria
La struttura secondaria di una proteina è la sua struttura
tridimensionale locale.
Per metterla in evidenza procedete così:
Chiudete e riaprite MGL Tools.
Aprite 1uv6_tut, deselezionate il cerchietto rosso
nella colonna L per non vedere più gli amminoacidi.
Selezionate il cerchietto nella colonna R per rappresentare
la proteina con un nastro (Ribbon).
Per colorare la struttura cliccate il triangolo nella colonna Cl
e scegliete By secondary structure, come in figura 6
Nella finestra Tools impostate il valore minimo di fog
intorno a 150 (un po' minore di quello massimo, 173) per vedere la molecola
ben illuminata.
Otterrete la struttura mostrata qui sotto, in figura 7.

I colori, giallo, rosso e bianco, rappresentano le tre
principali strutture secondarie delle proteine.
giallo = beta pieghe (beta strand)
rosso = alfa elica (alpha helix)
bianco (e azzurro) = avvolgimento casuale (random coil)
|
|
|
|
|
Struttura beta pieghe.
Esaminiamo per prima la struttura beta. Ogni
legame peptidico si trova adagiato su un foglietto e rivolge il calbonile
da un lato e il legame NH dal lato opposto. Ogni foglietto è
allineato ai successivi formando una specie di fisarmonica che dà
il nome alla struttura (beta pieghe).

Dato che l'ossigeno dei calbonili e l'idrogeno
degli NH sporgono lateralmente verso l'esterno, i tratti con avvolgimento
beta pieghe sono stabilizzati dalla formazione di legami idrogeno con
un'altra catena beta pieghe vicina. Per questo motivo i tratti beta
pieghe non si trovano mai da soli, ma sono affiancati uno all'altro
e, inoltre, sono prevalentemente antiparalleli (osservate la
posizione dei gruppi COOH
termiali qui sotto o la direzione delle frecce gialle in figura 8).

Nella proteina che stiamo studiando, il recettore
dell'acetilcolina, le catene beta sono organizzate per formare una struttura
a tubo come è evidente in figura 7. Le cinque subunità
formano quindi cinque strutture a tubo che, allontanandosi o avvicinandosi
tra loro, possono aprire o chiudere il poro per gli ioni sodio (visibile
al centro in figura 1).
I legami idrogeno tra le catene beta possono essere messi in evidenza
seguendo la procedura descritta qui sotto.
Individuiamo un punto nella subunità C nel quale ci siano almeno
tre segmenti beta pieghe affiancati come in figura 8.
Selezionando a turno gli amminoacidi delle varie zone beta pieghe (evidenziati
da una cornice gialla) si possono individuare gli amminoacidi di questi
tre segmenti: si trovano nei tratti 72-77 e 101-114.
Selezionate uno per uno questi 20 amminoacidi e poi salvateli
col nome 1uv6_beta.pdb.
La sequenza 101-114 è costituita da un stesso tratto di catena
che si ripiega su se stesso per creare un appaiamento antiparallelo
di strutture beta pieghe che è chiamato a "forcina".
Chiudete e riaprite MGL Tools.
Aprite il piccolo tratto di proteina appena salvato 1uv6_beta.pdb
(potete anche scaricarlo qui).
Selezionate la vista Balls and Sticks (colonna B)
Nella finestra Tools impostate il valore minimo di fog
intorno a 50 (un po' minore di quello massimo, 57) per vedere meglio
la struttura più lontana da voi.
Aggiungete gli idrogeni alla struttura, dato che mancano nel
file PDB originale.
Selezionate: Edit / Hydrogens / Add e poi cliccate OK.
Ora scegliete: Edit / Hydrogens / Merge non Polar e cliccate
Continue.
Nella struttura rimangono visibili solo gli idrogeni polari (figura
9). Si vede chiaramente che gli idrogeni degli N-H di una catena
sono rivolti verso gli ossigeni dei carbonili C=O della catena di fronte.
Cliccate ora: Hydrogen Bonds / Build / Set Parms + Build. Compare
una finestra nella quale dovete cliccare Specify two sets e così
potete scegliere quali legami idrogeno costruire.
Cliccate AtomSet List e scegliete backbone+h come è
mostrato in figura 10.
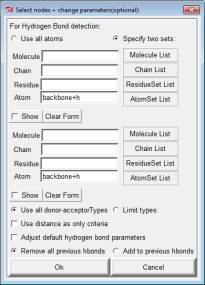 figura 10
figura 10
Dopo aver premuto OK, compare una finestra che
annuncia che sono stati individuati 9 legami idrogeno.
Cliccate ora Hydrogen Bonds / Display / As Spheres e vedrete
i 9 legami idrogeno rappresentati con piccole sfere verdi come in figura
11. Ogni singolo legame idrogeno può essere acceso o spento
cliccando sulla sua stringa nella finestra di dialogo.
Accendete infine la rappresentazione Ribbon nella colonna
R per vedere meglio la struttura beta pieghe collegata dai legami
idrogeno come in figura 12.
Notate che le catene R degli amminoacidi sporgono alternativamente
sopra e sotto il piano del foglietto beta
La struttura beta pieghe delle proteine, a causa dei legami idrogeno
intercatena, è appiccicosa, cioè può aggregarsi
ad altre catene proteiche con struttura beta pieghe come se fossero
striscie di nastro adesivo. In casi eccezionali questa appiccicosità
può formare aggregati proteici sempre più grandi che non
sono più degradabili dagli enzimi proteolitici delle cellule.
Il morbo di Alzheimer, per esempio, è dovuto all'aggregazione
di un piccolo peptide che inizialmente è avvolto con struttura
ad alfa elica, ma può riarrangiarsi ed assumere struttura beta
a pieghe. Questo peptide è noto come peptide beta amiloide
perchè, quando assume la conformazione beta, può produrre
degli aggregati che continuano a crescere fino a rompere le cellule
nervose in cui si trovano per poi fondersi con quelli vicini formando
dei grumi così grandi che sono visibili al microscopio ottico
e ricordano le sferette di amido delle cellule vegetali. Intere zone
del cervello risultano danneggiate in modo permanente.
|
|

fig. 13

fig. 14

fig. 15

fig. 16
|
|
Struttura ad alfa elica
Nell'alfa elica, la proteina si avvolge su se stessa
con spire destrorse dalle quali le catene R degli amminoacidi
sporgono a raggiera.
L'alfa elica è stabilizzata da legami idrogeno interni
alla catena, paralleli all'asse, che legano tra loro amminoacidi
che si trovano su spire successive. Ponendo in alto il lato N-terminale,
gli ossigeni dei carbonili C=O sono rivolti in basso e formano legami
idrogeno con gli idrogeni dei gruppi NH rivolti verso l'alto. Le strutture
ad alfa elica, quindi non sono appiccicose e non tendono a formare aggregati.
Chiudete e riaprite MGL Tools.
Aprite 1uv6_tut.pdb, togliete la rappresentazione a linee (colonna
L) e selezionate, per la catena C, la rappresentazione Ribbon (colonna
R), infine colorate il nastro (colonna Cl) a seconda della struttura
secondaria (figura 6).
La catena C contiene una sola alfa elica (rossa) mostrata nella figura
qui a fianco (figura 13).
Individuate gli amminoacidi dall'alfa elica (evidenziati da una cornice
rossa): si trovano nel tratto tra ARG-3 e GLN-12.
Selezionate uno per uno i 13 amminoacidi da ASP-2 a SER-14 e
poi salvateli col nome 1uv6_alfa.pdb.
Chiudete e riaprite MGL Tools.
Aprite il piccolo tratto di proteina appena salvato 1uv6_alfa.pdb
(potete anche scaricarlo qui).
Selezionate la vista Balls and Sticks (colonna B).
Nella finestra Tools regolate i valori minimo e massimo di
fog per vedere ben illuminata la struttura.
Selezionate la rappresentazione Ribbon (colonna R) e coloratela in base
alla struttura secondaria.
Otterrete l'immagine di figura 14 nella quale sono ben visibili
le catene R degli amminoacidi che sporgono radialmente
I legami idrogeno intracatena possono essere messi in evidenza seguendo
la procedura descritta qui sotto.
Togliete la rappresentazione Ribbon (colonna R)
Aggiungete gli idrogeni alla proteina.
Selezionate: Edit / Hydrogens / Add e poi cliccate OK.
Ora scegliete: Edit / Hydrogens / Merge non Polar e poi cliccate
Continue.
Nella struttura rimangono visibili solo gli idrogeni polari. Osservate
che gli idrogeni degli N-H della catena ad alfa elica sono rivolti verso
l'alto e si avvicinano agli ossigeni dei carbonili C=O della spirale
soprastante che sono tutti rivolti in basso.
Cliccate ora: Hydrogen Bonds / Build / Set Parms + Build. Compare
una finestra nella quale dovete cliccare Specify two sets e così
potete scegliere quali legami idrogeno costruire, cliccate AtomSet
List e scegliete backbone+h come è mostrato in
figura 10.
Dopo aver premuto OK, compare una finestra che annuncia che sono stati
individuati 8 legami idrogeno.
Cliccate ora Hydrogen Bonds / Display / As Spheres e vedrete
8 legami idrogeno rappresentati con piccole sfere verdi come in figura
15. Oni singolo legame idrogeno può essere acceso o spento
cliccando sulla sua stringa nella finestra di dialogo.
Accendete infine la rappresentazione Ribbon (colonna R) per
vedere la struttura ad alfa elica con i legami idrogeno intracatena
come in figura 16.
Si nota molto bene che i legami idrogeno mantengono compatte le varie
spire. Se la proteina è posta in acqua calda, i legami idrogeno
si rompono e l'alfa elica si allunga fino a scomparire.
Questo è quello che succede quando
si lavano i capelli con acqua calda, l'alfa elica delle proteine del
capello (alfa cheratina) si allenta e le fibre si allungano. I capelli
appena lavati, infatti, sono un po' più lunghi, ma tornano della
lunghezza normale quando si asciugano e i legami idrogeno si riformano
ripristinando la struttura ad alfa elica.
|
|
|
|
|
Struttura Terziaria
La struttura terziaria di una proteina è
la struttura tridimensionale complessiva di una catena proteica.
In figura 17 è mostrata la struttura terziaria della catena C.
Ogni catena proteica ha la propria struttura tridimensionale che è
la più stabile tra tutte quelle possibili, cioè corrisponde
ad un minimo di energia. La proteina assume spontaneamente questa configurazione
a mano a mano che viene sintetizzata, a meno che non sia troppo piccola
come l'insulina (che viene sintetizzata con una catena più
lunga che le consente di avvolgersi e poi viene tagliata) o molto grande
e allora ha bisogno di un aiuto da parte dei Chaperon, grosse
proteine a forma di barile nella cui cavità le proteine trovano
un ambiente ideale per avvolgersi, privo di interferenze.
In generale gli amminoacidi apolari tendono ad occupare la parte
più interna della proteina in modo da interagire tra loro
piuttosto che con il solvente acquoso nel quale la proteina è
immersa. Gli amminoacidi polari, invece, si rivolgono all'esterno,
verso l'acqua. Questo comportamento ricorda quello dei saponi che si
organizzano in micelle con le code apolari rivolte al centro e le teste
polari rivolte all'esterno.
Le proteine di membrana, come il recettore dell'acetilcolina, fanno
eccezione a questa regola e hanno una zona apolare in superficie, quella
che deve essere immersa nello strato di fosfolipidi della membrana cellulare.
Per ottenere l'immagine di figura 17 procedete così:
Chiudete e riaprite MGL Tools.
Aprite 1uv6_tut.pdb e selezionate la catena D, quindi
cancellatela con Edit / Delete / Delete Selected Atoms.
Nella catena C (unica rimasta) deselezionate la rappresentazione
a linee (colonna L) e selezionate la rappresentazione Ribbon
(colonna R). Colorate il nastro (colonna Cl) con l'opzione By
Secondary Structure.
Andate sul legando CCE-1206 e selezionate la rappresentazione
a Sfere (colonna C), quindi coloratela (colonna Cl) scegliendo
geometria cpk e By atom type. Infine aggiustate i valori
di fog nel menù Tools per vedere chiaramente la
struttura (figura 17).
Per valutare la polarità della catena C,
deselezionate Ribbon e rappresentate la proteina con sfere (colonna
C), quindi coloratela (colonna Cl) scegliendo cpk e poi By
polarity (David Godsell).
Otterrete l'immagine di figura 18 dove si vede che la zona
centrale della catena (quella immersa nella membrana) è relativamente
apolare (colori tenui), mentre l'estremità in alto
e la porzione in basso (che sporgono dalla membrana) sono molto
più polari, infatti abbondano amminoacidi basici (con
sfere blu) e acidi (con sfere rosse).
Si arriva alla stessa conclusione rappresentando la catena C con la
Superficie Molecolare (colonna MS) e poi colorandola (colonna
Cl) scegliendo cpk e poi By polarity (David Godsell).
Si ottiene l'immagine di figura 19.
|
|

fig. 20

fig. 21

fig. 22
figura 23
|
|
Struttura Quaternaria
Le proteine composte da più catene (subunità)
possiedono una struttura quaternaria cioè una struttura tridimensionale
complessiva delle subunità che descrive come queste si incastrano
una nell'altra per formare una struttura proteica funzionale.
Una proteina con struttura quaternaria può essere paragonata
ad un motore che è composto da diverse parti che devono essere
incastrate insieme correttamente per poter funzionare.
Il recettore dell'acetilcolina è una proteina con struttura
quaternaria formata da cinque subunità.
Scaricate il file 1uv6.pdb che
contiene la proteina completa e mettetelo nella cartella Acetilcolina.
Chiudete e riaprite MGL Tools.
Aprite 1uv6.pdb e eliminate le molecole di acqua con Edit
/ Delete water.
Deselezionate la rappresentazione a linee (colonna L) e selezionate
la rappresentazione Ribbon (colonna R). Colorate il nastro
(colonna Cl) con l'opzione By Secondary Structure. Aggiustate
il valore minimo di fog nel menù Tools ad un valore
un pò sotto quello massimo e otterrete l'immagine mostrata in
figura 20.
Per differenziare le 5 catene e mettere in evidenza i due legandi (carbammilcolina),
procedete così:
Su 1uv6.pdb Colorate il nastro (colonna Cl) con l'opzione By
Chain.
Aprite l'elenco dei residui della catena C, andate sul legando
CCE-1206 e selezionate la rappresentazione a sfere (colonna
C).
Aprite l'elenco dei residui della catena D, andate sul legando
CCE-1206 e selezionate la rappresentazione a sfere (colonna
C).
Otterrete l'immagine mostrata in figura 21.
Questo recettore dell'acetilcolina si trova legato ai due legandi e
quindi è nella conformazione col poro aperto. Per valutare meglio
le dimensioni e la polarità del poro potete rappresentare la
superficie molecolare della proteina colorata in base alla polarità.
Togliete la rappresentazione Ribbon alle cinque catene A, B,
C, D, E una per una.
Su 1uv6.pdb cliccate la rappresentazione della Superficie
Molecolare (colonna MS) e poi coloratela (colonna Cl) scegliendo
MSMS-Mol e poi By polarity (David Godsell).
Si ottiene l'immagine di figura 22.
Notate che la porzione esterna del poro è abbastanza apolare,
cioè è colorata con colori più tenui, mentre l'imboccatura
e l'interno del poro sono zone ricche di amminoacidi basici (blu) e
acidi (rossi) come si vede nella figura 23 qui sotto. Nel pori,
infatti devono transitare ioni sodio.

Autore: prof Mauro Tonellato
|
|
|
|
|
Chimica al Computer
PianetaChimica home
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



















